Article Text

Statistics from Altmetric.com
Chronic rhinosinusitis (CRS) is a heterogenous airways disease characterised by inflammation of the upper airways and sinuses that persists for at least 12 weeks. CRS is frequently divided into two endotypes based on the presence or absence of nasal polyps (NPs): CRS with NPs (CRSwNP) and CRS without NPs (CRSsNP). Inflammatory patterns in the two groups are different and studies indicate that CRSsNP is dominated by a type 1 inflammatory profile associated with increased interferon (INF)-γ and tumour necrosis factor (TGF)-β expression and neutrophilic inflammation.1 By comparison, type 2 inflammatory responses characterised by interleukin (IL) 4, IL-5, IL-13 cytokine expression and eosinophilic inflammation are features of CRSwNP.1 This categorisation may, however, be oversimplified as recent studies show that this may be subject to racial and regional differences.2 ,3 While the majority of Caucasian patients sampled in the USA and Europe have a pronounced infiltration of eosinophils and expression of IL-5 in the NPs, patients with CRSwNP in East Asian countries including Japan, Korea and China, exhibit a mixed inflammatory profile. About half the Asian patients with CRSwNP exhibit an eosinophilic chronic rhinosinusitis (ECRS) while the rest have a non-eosinophilic more neutrophilic or mixed inflammatory chronic rhinosinusitis (non-ECRS) characterised by type 1 dominant inflammation. Although a number of hypotheses have been proposed regarding the pathogenesis of CRSwNP, the precise molecular mechanisms remain unclear and likely lie beyond rudimentary clinical classifications of CRS. An emphasis on defining CRS endotypes based on distinct functional or pathobiological mechanisms of disease may be more effective at identifying patient groups that would respond to therapeutic interventions targeting specific proinflammatory mediators or infectious agents that trigger the disease pathology.
The inflammatory profile of CRSsNP is largely type 1, although there are discrepancies over which isoforms of TGF-β are increased per se and in what racial populations of CRSsNP, INF-γ is elevated. Since CRSsNP is diagnosed by the absence of NPs, it is by definition heterogenous and thus the outcome of these studies is heavily influenced by the population sampled.4 Accurate subclassifications will be necessary to determine more accurately the inflammatory pattern in each group.
Inflammation in CRSwNP is characterised by type 2 inflammation and increased tissue eosinophilia compared with CRSsNP or control sinus mucosa. Type 2 cytokines, IL-5 and IL-13, can be produced by several immune cells including T helper two cells, mast cells and group 2 innate lymphoid cells (ILC2). IL-5 promotes local differentiation of eosinophil-lineage committed progenitors and increased activation and prolonged survival of mature eosinophils, IL-13 activates macrophages, B cells and epithelial cells to release chemoattractants that induce recruitment of eosinophils and Th2 cells and IgE mediated reactions and tissue remodelling.1 In contrast to CRSsNP, fibrin deposition in the submucosa of NPs is especially high and thought to represent a key event in the tissue remodelling process of CRSwNP.4 Several studies have now shown that the inflammatory profile of CRSwNP varies among racial groups although the exact aetiology of these differences remains to be clarified. It is undeniable that this may also reflect a lack of international consensus for defining tissue eosinophilia. While some groups define eosinophilia as an absolute number of eosinophils (five to eight) per random high powered field,3 ,5 ,6 others use a cut-off percentage of eosinophils (5–15%) for a number of high power fields counted in areas of greatest cellularity.7 ,8
It has become evident that disruption of the mucosal interface in response to environmental and microbial stimuli or by cellular damage may drive local inflammatory responses. IL-33 is a recently described airway epithelial-derived cytokine recognised as a key activator of innate and adaptive cells that drive type 2 immune responses, contributing to tissue homoeostasis and response to environmental stresses (figure 1). IL-33 was identified as a ligand for the orphan IL-1 family receptor, suppression of tumorigenicity 2 (ST2; also known as IL-1RL1, T1, IL-33R) expressed on monocytes, mast cells, eosinophils, Th2 cells and ILC2. IL-33 exerts its cytokine activity by binding to a heterodimer of ST2 and co-receptor, IL-1 receptor accessory protein. IL-33 is a chromatin-associated nuclear cytokine and as such is expressed constitutively at high levels in the nuclei of various human and mouse tissues at steady state, including endothelial, epithelial and fibroblast-like cells.9 This nuclear expression can be further increased during inflammatory conditions, viral infection and cigarette smoke. IL-33 does not contain a signal sequence and therefore is not secreted as a conventional cytokine. Instead, it functions as an endogenous danger signal or nuclear alarmin that is released after cell injury to alert the immune system of damage during trauma or infection. Once released the bioactive isoforms of IL-33 can stimulate type 2 cytokine production by ILC2 and IL-13 production by macrophages which in turn promote a maladaptive TH2 inflammatory response that is IgE-independent and may explain the clinical observation that many patients with CRSwNP are non-allergic (figure 1).4

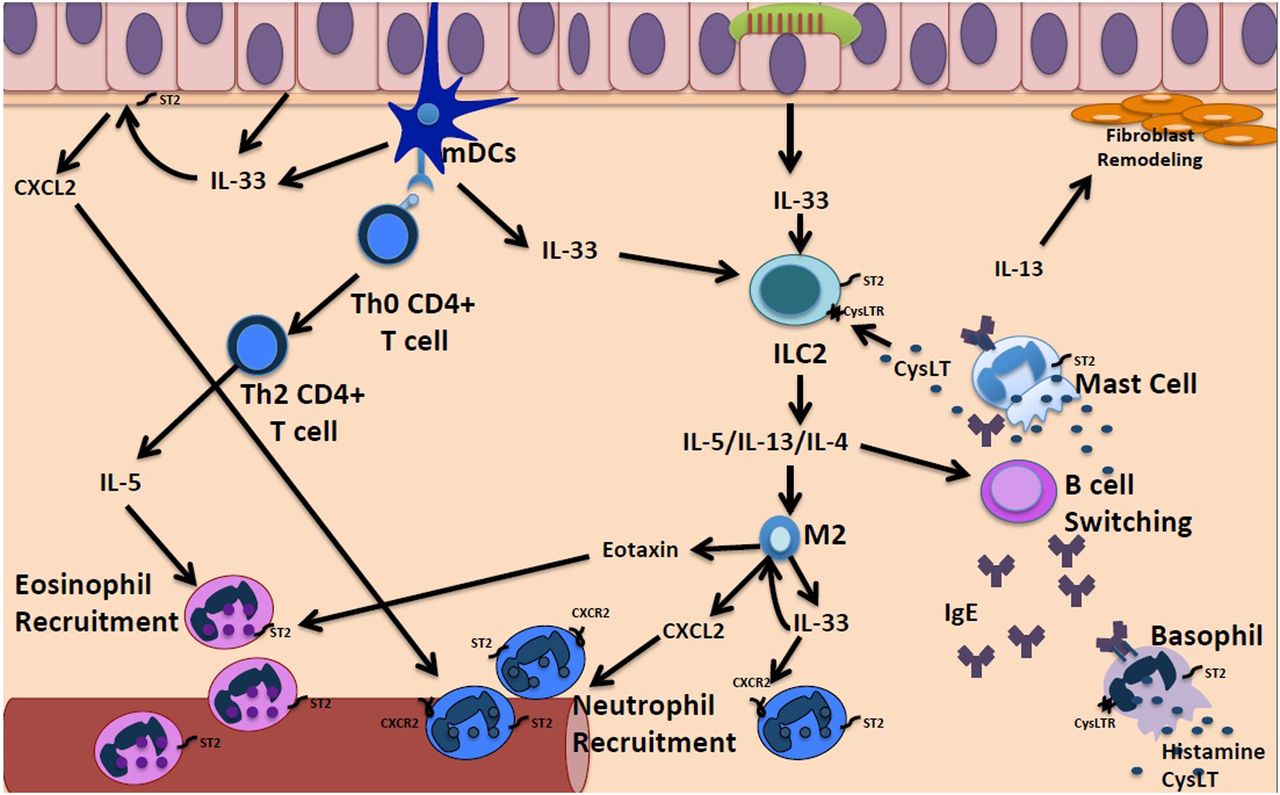{kind=link}
Potential mechanisms for amplification of neutrophilic inflammation in CRSwNP. Several factors including fungi, proteases, bacteria viruses and allergens can activate nasal epithelial cells and resident myeloid dendritic cells (mDCs) to produce alarmin cytokines including IL-33 which drive maladaptive innate type 2 inflammatory responses. IL-33 stimulates resident group 2 innate lymphoid to secrete IL-4, IL-5 and IL-13 in an allergen non-specific manner which promotes dysregulated type 2 immune responses. mDCs activation also promotes naive CD4+ T cell differentiation into Th2 CD4+ T cells, which then further release type 2 cytokines. IL-4 and IL-13 induce B cell isotype switching and leads to IgE-mediated reactions through activation of basophils and mast cells. Nasal mast cells can also be activated directly by IL-33, leading to downstream release of cysteinyl leukotrienes (CysLT), which activate ILC2s, and IL-5 and IL-13, which further contribute to downstream type 2 inflammation. IL-5 production from activated Th2 CD4+ T cells and ILC2s promote eosinophil recruitment, as well as eotaxin produced by nasal epithelial cells and M2 macrophages. In addition, IL-33 which can indirectly promote neutrophilia by activating resident proinflammatory cells such as M2 macrophages to secrete (1) chemoattractants that recruit neutrophils including CXCL2 or (2) produce more IL-33 that prevents downregulation of CXCR2, the receptors for CXCL2 on neutrophils. ILC2, group 2 innate lymphoid cell; ST2, suppression of tumorigenicity 2.
IL-33 expression has been shown to be increased in cultures of sinonasal epithelial cells in recalcitrant CRSwNP, and it is further enhanced in a bacteria associated molecular pattern.10 In addition, other studies have shown that expression of ST2 is increased in the ethmoid sinus mucosa of CSRwNP compared with CRSsNP.11 Similarly, in subjects with CRS with eosinophilic NPs, increased ST2 levels were detected at the mRNA and protein levels, compared with non-eosinophilic CRS and normals. The levels of IL-33 mRNA were however not different between these groups12 and correlation of the cytokine expression to various proinflammatory cell types was not investigated.
The study by Kim et al, published in Thorax, assessed lL-33 expression at the protein and message level with correlations to differential groups of proinflammatory cells and cytokine expression in Korean subjects with CRSwNP and CRSsNP versus normal control.13 This study found that IL-33 expression is increased in two mucosal sites: NP and uncinate process from CRS and that IL-33 immunostaining is colocalised to CD68+ macrophage. Although several studies have shown that CD45+ haemopoietic cells can be induced to express IL-33 under inflammatory conditions, the levels observed after stimulation are considerably lower than those expressed by tissue-resident and structural cells. Therefore the exact physiological relevance of IL-33 production by macrophages in CRSwNP remains to be clarified. Further studies with bone marrow chimaeras and cell-specific knockout approaches may be required to determine the relative importance of the different cellular sources of IL-33.
The study by Kim et al also found that total tissue extracted IL-33 protein levels in NP tissue correlated with elastase+ neutrophil numbers and Th1 and Th17 cytokine levels but had an inverse relation to major basic protein+ eosinophils and IL-5. It is known that full length IL-33 is activated by mast cell proteases and neutrophil-derived elastase enzymes, so it is not clear whether IL-33 secretion promotes neutrophilia or vice versa. However, the antibodies used in the study recognise the overlap region between full length IL-33 and the bioactive secreted isoforms of IL-33 supporting the former proposal. In addition, IL-33 has previously been shown to promote tissue neutrophilia albeit indirectly by preventing downregulation of CXCR2 thus supporting the findings in this study that IL-33 stimulated upregulation of neutrophil-recruiting chemokines, CXCL2, which may in turn promote tissue-homing of these cells.14 Kim et al did not, however, analyse the subset(s) of IL-33-responsive innate immune cells involved in reducing neutrophil recruitment in chronic nasal mucosa inflammation. To understand the drivers of chronic inflammation in CRS it may be important to investigate this further particularly in view of the number of CRSwNP that were non-atopic (>70%) in this study. In addition, this paper did not measure levels of soluble ST2 which is known to be a negative decoy for IL-33.15 Assessing for variations between the subject groups may have provided information on other treatment options in addition to anti-IL-33.
In a mouse model of CRS exposed to ovalbumin/Staphylococcal enterotoxin B that generated a mixed inflammatory infiltrate, Kim et al showed that anti-IL-33 attenuated polyp formation, neutrophilia and CXCL2 levels but had no effect on eosinophil recruitment and IL-4 levels. As a note of caution, in assessing the role of IL-33 in animal models of disease, species-specific differences should be taken into account. For example, whereas in humans IL-33 is expressed constitutively by the lung airway epithelium, in mice it is expressed by alveolar type 2 pneumocytes.16 ,17 These differences may be important when extrapolating results from mouse models for therapeutic targeting of IL-33 in human patients.
In closing, Kim et al have shown that IL-33 expression is elevated in nasal tissues of patients with CRSwNP, and expression of this cytokine is associated with Th1/Th17 cytokines, neutrophil recruitment and remodelling. It is likely that IL-33 is one of the crucial mediators of pathogenesis in CRSwNP through neutrophil recruitment, but further study is required to understand if this is a promising target for the treatment of patients with all endotypes of CRSwNP or specific to pathobiological initiators of this upper airway inflammatory condition.
Footnotes
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Respiratory research