Article Text

Abstract
Until recently, three classes of medical therapy were available for the treatment of pulmonary arterial hypertension (PAH)—prostanoids, endothelin receptor antagonists and phosphodiesterase type 5 (PDE5) inhibitors. With the approval of the soluble guanylate cyclase stimulator riociguat, an additional drug class has become available targeting a distinct molecular target in the same pathway as PDE5 inhibitors. Treatment recommendations currently include the use of all four drug classes to treat PAH, but there is a lack of comparative data for these therapies. Therefore, an understanding of the mechanistic differences between these agents is critical when making treatment decisions. Combination therapy is often used to treat PAH and it is therefore important that physicians understand how the modes of action of these drugs may interact to work as complementary partners, or potentially with unwanted consequences. Furthermore, different patient phenotypes mean that patients respond differently to treatment; while a certain monotherapy may be adequate for some patients, for others it will be important to consider alternating or combining compounds with different molecular targets. This review describes how the four currently approved drug classes target the complex pathobiology of PAH and will consider the distinct target molecules of each drug class, their modes of action, and review the pivotal clinical trial data supporting their use. It will also discuss the rationale for combining drugs (or not) from the different classes, and review the clinical data from studies on combination therapy.
- Primary Pulmonary Hypertension
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Pulmonary arterial hypertension (PAH) is a chronic, potentially fatal disease characterised haemodynamically by increased mean pulmonary artery pressure ≥25 mm Hg, normal pulmonary artery wedge pressure ≤15 mm Hg and elevated pulmonary vascular resistance (PVR) >3 Wood Units. PAH is caused by progressive remodelling of the pulmonary vasculature by cell proliferation and fibrosis, occluding the blood vessels and ultimately leading to right ventricular failure and death.1–3 The vascular pathology of PAH results at least in part from endothelial cell dysfunction, accompanied by impaired signalling in several pathways.4–9 Despite advances in current therapies for PAH, there is still a significant unmet medical need, as the mortality of patients with PAH remains high.10–12
Until recently, three classes of medical therapy were available for the treatment of PAH targeting three dysfunctional pathways—prostanoids, endothelin receptor antagonists (ERAs) and phosphodiesterase type 5 (PDE5) inhibitors. With the approval of the soluble guanylate cyclase (sGC) stimulator riociguat,13–16 a new, fourth class of therapy has become available, targeting the same pathway as PDE5 inhibitors. The treatment algorithm discussed at the 5th World Symposium on Pulmonary Hypertension (PH)17 recommends the use of all four drug classes to treat PAH (table 1), as do the recently published CHEST guidelines on pharmacological therapy for PAH in adults (table 2).18 However, there is a lack of comparative data for these therapies; therefore, an understanding of the mechanistic differences between these agents and the clinical data sets supporting their use is critical when making treatment decisions.
Treatment recommendations from the 5th World Symposium on Pulmonary Hypertension17
Summary of CHEST/American College of Chest Physicians guidelines for pharmacological treatment of PAH18
Inter-individual differences mean that patients can respond differently to the same PAH treatment. Monotherapy may be adequate for some patients, while others may only improve when treated with a combination of two or three different classes of drugs. Although it is unclear whether these patients are benefiting from combination therapy, or whether the use of more than one drug is increasing the patient's chances of receiving the treatment to which they are most responsive, for patients who receive combination therapy, it is important to consider alternating or combining compounds with different molecular targets. Combination therapy is often used to treat PAH and it is important, therefore, that physicians understand how the modes of action of these drugs might result in additive, synergistic or antagonistic interactions which could lead to differences in safety and efficacy.
This review will give a comprehensive overview of the available approved medical treatments for PAH, describing how each drug class targets the complex pathobiology of PAH, and will consider the distinct target molecules of each drug class. We will also discuss the rationale for combination therapy and review the clinical evidence for the use of combination therapy.
Currently approved therapies and their molecular targets
The modes of action of the approved classes of therapy for PAH are illustrated in figure 1. Key positive clinical trial data for each therapy are summarised in tables 3 and 4.
Positive randomised controlled trials of PAH therapies
Long-term extension studies
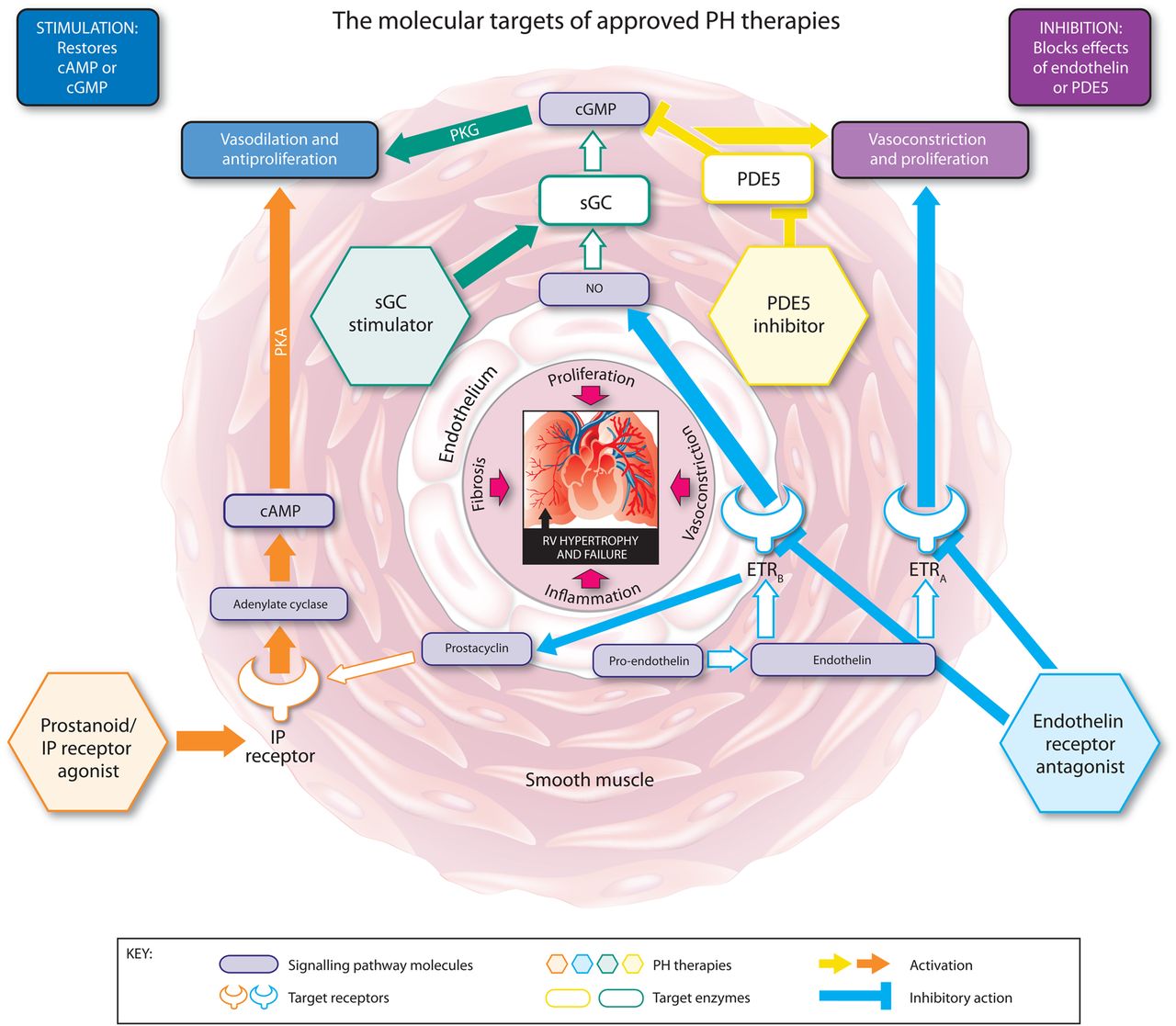
The molecular targets, signalling pathways, and modes of action of approved pulmonary hypertension (PH) therapies. cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; ETRA, endothelin receptor A; ETRB, endothelin receptor B; IP, prostacyclin; NO, nitric oxide; PDE5, phosphodiesterase type 5; PKA, phosphate kinase A; PKG, cGMP-dependent protein kinase; sGC, soluble guanylate cyclase.
Prostanoids: stimulation of adenylate cyclase through prostacyclin (IP) receptor agonism
Prostaglandin I2 (PGI2) or prostacyclin is a potent pulmonary vasodilator synthesised in the endothelium.36 Its main target, the IP receptor, is expressed in the vascular smooth muscle cell (VSMC) layer of the pulmonary vasculature37–39 as well as other tissues. Activation of the IP receptor results in the conversion of ATP to cyclic AMP (cAMP) which increases protein kinase A (PKA) activity, leading to downstream effects including vasodilation. Prostacyclin has antithrombotic, antiproliferative, antimitogenic and immunomodulatory properties.38 ,40–42 Prostacyclin synthase and prostacyclin metabolites are reduced in the pulmonary vasculature and blood serum of patients with PH.43 ,44 The prostanoid drugs are synthetic analogues of prostacyclin, designed to substitute the endogenous prostacyclin that is lacking in these patients.
Epoprostenol
Epoprostenol is a synthetic salt of PGI2, and was the first prostanoid to be tested in patients with PAH.19 It is approved in the USA, Europe and several other regions for treatment of PAH in patients in WHO functional class (WHO FC) III and IV, who do not respond adequately to conventional therapy. Epoprostenol is unstable, with a half-life of <5 min, and is administered intravenously via an indwelling central venous catheter connected to an infusion pump. As such, meticulous hygiene must be observed due to the risk of catheter site infection. Interruption of drug supply can result in severe and potentially fatal rebound PH.45 ,46 In 2008, a novel formulation was introduced (Veletri, Actelion Pharmaceuticals Ltd, Allschwil, Switzerland) that is stable at room temperature for up to 24 h, eliminating the need for ice packs to cool the pump. Details of the randomised trial of epoprostenol are listed in table 3.
Iloprost
Iloprost is a carbacyclin analogue of PGI2 that has improved stability at room temperature and a longer half-life (20–30 min) compared with epoprostenol. It can be administered intravenously or by inhalation. Inhaled iloprost is approved in the USA for patients with PAH in WHO FC II or III, and in Europe and several other countries for patients with PAH in WHO FC III. Details of the AIR study of inhaled iloprost are listed in table 3. Intravenous iloprost treatment has been evaluated in several open-label uncontrolled clinical studies, however not in a randomised controlled clinical trial, and is only approved in New Zealand for PAH.
Beraprost
Beraprost is a prostanoid with a half-life of 35–40 min that is administered orally. Although treatment with beraprost initially improved 6 min walking distance (6MWD) (ALPHABET study, table 3),24 long-term follow-up showed that the improvement was not sustained.47 Beraprost is approved in Japan, South Korea and some other South-East Asian countries such as Indonesia for treatment of PAH in patients in WHO FC III.
Treprostinil
Treprostinil is an analogue of PGI2 which has a longer half-life (up to 4 h) and improved stability compared with epoprostenol. It can be administered via subcutaneous, intravenous, inhaled and oral routes. Subcutaneous treprostinil is approved in the USA for the treatment of patients with PAH in WHO FC II–IV, and in Europe for patients with PAH in WHO FC III. Intravenous treprostinil should be reserved for patients who are intolerant of the subcutaneous route, patients with a preference for intravenous administration, or in whom the risks are considered warranted. Inhaled and oral treprostinil are only approved in the USA, and oral treprostinil is only approved in treatment-naïve patients with PAH, whereas inhaled treprostinil is only approved for use in combination therapy. Details of the randomised trials of subcutaneous, inhaled (TRIUMPH I) and oral treprostinil (FREEDOM-M) are listed in table 3.
Selexipag
Selexipag is a selective, non-prostanoid IP receptor agonist. It is rapidly metabolised to an active form with a half-life of 7.9 h, and is administered orally. It is not yet approved in the USA or Europe. Following a phase II study in 43 patients,48 the recent GRIPHON phase III event-driven study in 1156 patients indicated that treatment with selexipag significantly decreased the relative risk of a morbidity/mortality event compared with placebo. At the time of writing this article, the results of the GRIPHON trial have only been presented in abstract form.49 ,50
Blockade of endothelin receptors
Endothelin 1 (ET-1) is a potent vasoconstrictor and promoter of pulmonary artery smooth muscle cell proliferation51 which is upregulated in PAH.52 There are two ET-1 receptor subtypes: endothelin receptors A (ETAR) and B (ETBR). ETAR is highly expressed in VSMCs and contributes to ET-1-induced contraction and proliferation of VSMCs in PAH.53 ,54 ETBR is predominantly expressed in vascular endothelial cells where activation with ET-1 promotes vasodilation through accelerated production of PGI2 and nitric oxide (NO) and clearance of ET-1,55–57 but is also expressed in VSMCs, where it has vasoconstrictive and proliferative actions.55 ET receptors are also upregulated in PAH.51 The dual ERAs bosentan and macitentan, and the selective ETAR antagonist ambrisentan bind and block the ET receptors, preventing ET-1-mediated responses.
Bosentan
Bosentan is a dual ERA, administered orally. It is approved for the treatment of patients with PAH in WHO FC II–IV in the USA and several other regions, and in patients in WHO FC II–III in Europe. It may be associated with increased levels of liver transaminases; therefore patients are started on a low dose, which is up-titrated to the therapeutic dose as long as liver transaminase levels remain normal. Monthly monitoring of liver transaminases is mandatory for patients taking bosentan.58 Details of the BREATHE-1 and EARLY studies are given in table 3.
Ambrisentan
Ambrisentan is a selective ETAR antagonist with a longer half-life than bosentan (9 h vs 5.4 h, respectively), administered orally. It is approved for the treatment of patients with PAH in WHO FC II and III in the USA, Europe and several other regions. Theoretically, selective ETAR antagonists should be more effective than dual ERAs in that they only target the vasoconstrictive and mitogenic activities of ET-1–ETAR, while preserving the vasodilatory and antimitogenic effect of ET-1–ETBR.59 Despite its selectivity for ETAR, ambrisentan has not shown greater efficacy in non-comparative monotherapy clinical trials, compared with bosentan.60 This may be because the distribution or function of ETBR is altered in disease states, and thus dual receptor blockers appear to perform as well as those ERAs with a specific target.61 Unlike bosentan, ambrisentan was not associated with elevated liver toxicity,62 ,32 but peripheral oedema was reported more frequently compared with placebo.27 Details of the ARIES-1 and ARIES-2 trials are given in table 3, and the ARIES-E extension study is detailed in table 4.
Macitentan
Macitentan is a dual receptor ERA, administered orally. It is approved for the treatment of PAH for the long-term treatment of WHO FC II and III patients with PAH in the USA and Europe. Macitentan was not associated with liver toxicity.28 Decreased haemoglobin levels have been reported more frequently in patients receiving macitentan versus placebo in randomised controlled trials. Details of the SERAPHIN study are given in table 3.
Inhibition of phosphodiesterase-5
The PDE superfamily of enzymes inactivates and degrades cyclic guanosine monophosphate (cGMP) and cAMP, important vasoactive mediators that, via activation of cGMP-dependent protein kinase (PKG) and PKA, trigger vasodilation and inhibit proliferation (PKG can also be activated by increasing levels of cAMP). PDE5 is abundantly expressed in the VSMCs of the pulmonary vasculature and is upregulated in PAH in the VSMCs, and in right ventricle (RV) cardiomyocytes.63–66 The upregulation of PDE5 results in increased hydrolysis of cGMP. The PDE5 inhibitors sildenafil and tadalafil occupy the catalytic site on PDE5, blocking the degradation of cGMP.67 PDE5 inhibitors prevent cGMP degradation but are dependent on cGMP synthesis and the presence of NO.68 ,69 However, there is also evidence to suggest that the beneficial effects of PDE5 inhibition in PH are influenced by natriuretic peptide activity and the presence of natriuretic peptide-derived cGMP.70
Sildenafil
Sildenafil is an oral PDE5 inhibitor approved for the treatment of patients with PAH in WHO FC II–III in Europe and WHO FC II–IV in the USA. The details of SUPER-1 and SUPER-2, the long-term extension are given in tables 3 and 4.
Tadalafil
Tadalafil is an oral PDE5 inhibitor with a longer half-life than sildenafil, allowing once-daily dosing, rather than the three times daily dose that is indicated with sildenafil. It is approved in the USA, Europe and several other countries for the treatment of patients with PAH in WHO FC II–III. Details of the PHIRST-1 study and its long-term extension PHIRST-2 are given in tables 3 and 4.
Stimulation of sGC
sGC is expressed in the VSMCs of the pulmonary vasculature, platelets, RV and other tissues. When bound to NO, sGC catalyses the conversion of GTP to cGMP,71 ,72 promoting vasodilation and inhibiting smooth muscle proliferation, leucocyte recruitment, inflammation, fibrosis, platelet aggregation and vascular remodelling via several downstream mechanisms.73 The sGC stimulator riociguat is the first member of this novel class of therapeutics.69 ,73–75
Riociguat
Riociguat is an oral sGC stimulator with a dual mode of action. It sensitises sGC to endogenous NO by stabilising NO–sGC binding. Riociguat also directly stimulates sGC, independently of NO,73 ,69 resulting in increased generation of cGMP.73 ,69–76 Currently, riociguat is the only pharmacotherapy to be approved for the treatment of two PH indications, PAH and chronic thromboembolic PH (CTEPH).14–16 Riociguat is approved in the USA, Europe and several other regions for patients with group I PAH in WHO FC II or III; and for the treatment of patients with inoperable CTEPH, or persistent/recurrent PH after pulmonary endarterectomy in WHO FC II or III. Patients receiving riociguat have their dose individually adjusted up to the optimal dose over a period of 8 weeks, according to the patient's systolic systemic arterial blood pressure and signs or symptoms of hypotension.
Riociguat and PDE5 inhibitors act on separate targets within the NO signalling pathway. Combining them may increase the risk of amplifying their effects, leading to systemic hypotension. Therefore, the use of PDE5 inhibitors in conjunction with riociguat is contraindicated.14–16 ,77 Rare cases of haemoptysis have been reported with riociguat therapy in patients with PAH and CTEPH. Details of the PATENT-1 study and its long-term extension PATENT-2 are given in tables 3 and 4.
Combination therapy
Combining two or more classes of agent is a strategy that has been used successfully in the treatment of many other cardiovascular conditions such as systemic hypertension and heart failure.17 Combination therapy can be given as first-line treatment or by sequentially adding drugs to a patient's treatment regimen. The 2013 updated algorithm of PAH treatment from the 5th World Symposium on PH recommends sequential combination therapy, due to the high level of evidence of efficacy, in patients with PAH with an inadequate response to monotherapy, and possible first-line combination therapy in WHO FC III and IV patients with a lower recommendation.17 Several clinical trials have now assessed the effect of combination therapy in patients with PAH, with mixed results (see below). Several recent meta-analyses of the efficacy of combination therapy versus monotherapy, however, conclude that combination therapy can improve clinical outcomes, but has no proven effect on mortality.78–81 However, all acknowledge that more data are required from further studies.
First-line combination therapy
There are few data available on the use of combination therapy as a first-line treatment. The BREATHE-282 study investigated the effect of combined treatment with bosentan and epoprostenol versus epoprostenol and placebo in a small group of 33 patients with severe PAH (WHO FC III and IV) over 16 weeks, but did not demonstrate a significant difference between the two treatments in the primary endpoint of total pulmonary resistance. Three deaths were reported, all in the bosentan arm; however, investigators concluded that they reflected the severity and progressive nature of PAH and were not related to study treatment.
First-line combination therapy with ambrisentan and tadalafil was evaluated in the randomised, multicentre AMBITION study.83 ,84 This study had a dual placebo design, with one placebo group receiving ambrisentan monotherapy with placebo tadalafil, while the second placebo group received tadalafil monotherapy with placebo ambrisentan. With 610 patients enrolled, and followed up for up to 3.5 years,84 AMBITION was a considerably larger trial than the pioneer BREATHE-2 study. Significant improvements were seen in the composite primary endpoint of morbidity/mortality in the combination group compared with the pooled monotherapy arms, which were driven by differences in hospitalisation. Hierarchical testing of secondary endpoints showed significant improvements in N-terminal prohormone of brain natriuretic peptide (NT-proBNP), proportion of patients with satisfactory clinical response, and 6MWD. No important safety signals were observed. At the time of writing, the results of the AMBITION trial have only been presented in abstract form and have not yet been published.83
Sequential combination therapy
Several studies have evaluated the efficacy of adding a new therapy to a patient's existing PAH treatment regimen, with variable results (figure 2).

{kind=link}
{kind=link}
Combination therapy strategies tested in randomised controlled clinical trials. ERA, endothelin receptor antagonist; PDE5, phosphodiesterase type 5; sGC, soluble guanylate cyclase.
The addition of sildenafil or riociguat to prostanoid treatment has proved successful. The PACES study investigated the addition of sildenafil 80 mg three times daily (four times the approved dose) to the treatment of 267 patients with PAH already receiving epoprostenol.85 After 16 weeks of treatment, improvements were seen in 6MWD, mean pulmonary arterial pressure, cardiac output, time to clinical worsening (TTCW) and quality of life (QoL) compared with placebo. Adverse events (AEs) observed more often with sildenafil treatment were headache and dyspepsia.85 PATENT-131 investigated the efficacy and safety of riociguat as combination therapy and monotherapy. Of 443 patients, 222 were receiving ERAs (n=194) or non-intravenous prostanoids (n=28). Overall, after 12 weeks of treatment, 6MWD, PVR, NT-proBNP, WHO FC, TTCW, QoL and Borg dyspnoea score were significantly improved. Riociguat was effective in combination therapy and as monotherapy. The most common AEs that occurred during the study were headache, dyspepsia and peripheral oedema.31
Macitentan has been shown to be effective in patients already receiving either non-intravenous prostanoids or PDE5 inhibitors. The SERAPHIN study28 investigated whether long-term treatment with macitentan reduced a combined endpoint of morbidity or mortality in patients with PAH as either combination or monotherapy over 3 years.86 Of 742 patients treated, 471 were receiving either PDE5 inhibitors (n=454) or non-intravenous prostanoids (n=40). Overall, there was a significant reduction in the risk of a combined endpoint event in patients receiving macitentan compared with placebo, which was driven by deterioration in PAH. The risk of a combined endpoint event for patients receiving macitentan as monotherapy was lower than for patients receiving macitentan in combination therapy. However, patients receiving combination therapy showed a significant improvement in 6MWD, whereas patients receiving macitentan monotherapy did not. Nasopharyngitis, headache and anaemia were more common in patients receiving macitentan than in those receiving placebo.28
The efficacy of adding bosentan to other therapies has not been proven, despite its evaluation in several studies. In the phase IV, event-driven COMPASS-287 study, 334 patients receiving sildenafil for at least 12 weeks were randomised to either placebo or bosentan. The primary endpoint of time to the first morbidity/mortality event (defined as death, hospitalisation for PAH worsening or intravenous prostanoid initiation, atrial septostomy, lung transplant, or worsening PAH) was not met, with a non-significant observed risk reduction for morbidity/mortality. Improvements were observed in secondary endpoints 6MWD and NT-proBNP. The safety profile was consistent with that of bosentan, with no new signals observed.87
In a study design that was essentially a reversal of COMPASS-2, NCT0032329788 investigated the addition of sildenafil to the treatment of 103 patients with PAH already receiving bosentan. The trial did not meet its primary endpoint of a significant increase in 6MWD in patients treated with combined bosentan and sildenafil compared with patients treated with bosentan alone. The most common AEs in the sildenafil group were diarrhoea, headache and peripheral oedema.88
The addition of riociguat on top of bosentan therapy has been efficacious in PATENT-1.31 However, the STEP89 study investigating the addition of inhaled iloprost to bosentan treatment in patients with PAH (n=67) showed a numerically greater 6MWD in the inhaled iloprost plus bosentan group compared with bosentan alone at 12 weeks. While this trend did not reach statistical significance, there were significant improvements in WHO FC, TTCW, mean pulmonary artery pressure and PVR. The most frequently reported AEs in the iloprost group were consistent with the safety profile of prostanoids, and included headache, flushing and jaw pain.89
The PHIRST study30 investigated tadalafil as monotherapy or in combination therapy, with bosentan in 405 patients. After 16 weeks, improvement in 6MWD in patients receiving tadalafil as monotherapy was significant, whereas that of patients receiving combination therapy was not. The most common treatment-related AEs reported with tadalafil were headache, myalgia and flushing.30
COMPASS-390 was a study investigating the benefits of a stepped approach to combination therapy. One hundred treatment-naïve patients started receiving bosentan for 16 weeks. If they failed to achieve a 6MWD ≥380 m by week 16, they were given sildenafil in addition to bosentan. At the end of the trial, 31% of patients had achieved the ≥380 m threshold (16% on monotherapy and 15% on combination therapy). No statistical analysis is available. The most common reported AEs were peripheral oedema, dyspnoea, headache, dizziness, anaemia and abnormal liver function tests.90
There is some evidence that the delivery route of a drug may influence its efficacy in combination with other drugs. The TRIUMPH study21 investigated the effect of adding inhaled treprostinil to the therapy regimen of 235 patients with PAH who were receiving bosentan or sildenafil. After 12 weeks, there were significant improvements in 6MWD, QoL and NT-proBNP, but not in TTCW or WHO FC. AEs associated with treprostinil treatment were cough, headache and flushing.21 Conversely, the addition of oral treprostinil to similar background therapy as TRIUMPH was not successful. The FREEDOM-C study91 investigated the effect of adding oral treprostinil to the regimen of 350 patients receiving ERAs and/or PDE5 inhibitors. After 16 weeks, the primary endpoint, improvement in 6MWD, did not reach significance. AEs that resulted in study drug discontinuation included headache, nausea, diarrhoea, vomiting, worsening PH, extremity pain, chest discomfort and myalgia.91
The background therapy of a patient may also influence the choice of add-on treatment. For patients who are already receiving PDE5 inhibitors, riociguat is contraindicated. The phase II PATENT-PLUS77 study investigated the safety and clinical effect of riociguat in combination with sildenafil in 18 patients. After 12 weeks, combination therapy showed no favourable effects on exploratory clinical parameters, including haemodynamics and 6MWD. The primary endpoint of the trial, change from baseline in supine systolic blood pressure versus placebo, did not show a difference between riociguat and placebo. There were unfavourable safety signals (high rates of discontinuation due to hypotension and SAEs) with sildenafil plus riociguat in the long-term, single-arm, open-label phase of the study and no evidence of a positive benefit:risk ratio.77
Conclusions
The pathobiology of PAH is complex, and it is necessary for prescribing clinicians to have a thorough understanding of the mode of action of the drugs available for PAH and of the evidence base of various combinations. The approval of riociguat has introduced a new, fourth class of pharmacotherapy, targeting the NO–sGC–cGMP pathway with a different molecular target to previously approved PDE5 inhibitors. However, comparative studies of the approved classes of therapy are lacking. Although more evidence from trials of various therapies in combination is becoming available, the benefits of many therapy combinations remain inconclusive. The question of sequential versus first-line combination therapy is still under investigation, with further studies warranted. Combination therapy according to patient phenotype will also need to be guided by future studies. Despite the advances made in recent years, PAH remains an incurable disease and as such, there is still an unmet medical need for new therapies, possibly targeting alternative pathways.
References
Footnotes
Funding Editorial assistance, provided by Adelphi Communications Ltd (Bollington, UK), was sponsored by Bayer Pharma AG.
Competing interests MH has received consultancy fees from Actelion, Bayer, GSK, Novartis, Pfizer and Aires. He has received speaker fees from Actelion, Bayer, GSK, Novartis and Pfizer. H-AG has received consultancy fees from Actelion, Bayer, Ergonex, Gilead, GSK, Merck, Novartis and Pfizer. He has received speaker fees from Actelion, Bayer, Ergonex, Gilead, GSK, Novartis and Pfizer.
Provenance and peer review Not commissioned; externally peer reviewed.