Article Text

Abstract
Rationale Traffic-related air pollution has been shown to augment allergy and airway disease. However, the enhancement of allergenic effects by diesel exhaust in particular is unproven in vivo in the human lung, and underlying details of this apparent synergy are poorly understood.
Objective To test the hypothesis that a 2 h inhalation of diesel exhaust augments lower airway inflammation and immune cell activation following segmental allergen challenge in atopic subjects.
Methods 18 blinded atopic volunteers were exposed to filtered air or 300 µg PM2.5/m3 of diesel exhaust in random fashion. 1 h post-exposure, diluent-controlled segmental allergen challenge was performed; 2 days later, samples from the challenged segments were obtained by bronchoscopic lavage. Samples were analysed for markers and modifiers of allergic inflammation (eosinophils, Th2 cytokines) and adaptive immune cell activation. Mixed effects models with ordinal contrasts compared effects of single and combined exposures on these end points.
Results Diesel exhaust augmented the allergen-induced increase in airway eosinophils, interleukin 5 (IL-5) and eosinophil cationic protein (ECP) and the GSTT1 null genotype was significantly associated with the augmented IL-5 response. Diesel exhaust alone also augmented markers of non-allergic inflammation and monocyte chemotactic protein (MCP)-1 and suppressed activity of macrophages and myeloid dendritic cells.
Conclusion Inhalation of diesel exhaust at environmentally relevant concentrations augments allergen-induced allergic inflammation in the lower airways of atopic individuals and the GSTT1 genotype enhances this response. Allergic individuals are a susceptible population to the deleterious airway effects of diesel exhaust.
Trial registration number NCT01792232.
- Allergic lung disease
- Occupational Lung Disease
Statistics from Altmetric.com
Key messages
What is the key question?
Does freshly generated diesel exhaust augment effects of allergen exposure in the lower airways in atopic human subjects?
What is the bottom line?
We demonstrate that inhalation of diesel exhaust preceding segmental allergen challenge augments allergic inflammation in the lower airways of atopic individuals. Specifically, airway neutrophils, eosinophils, interleukin (IL) 5, IL-8, eosinophil cationic protein (ECP) and monocyte chemotactic protein (MCP)-1 were augmented.
Why read on?
This is the first study to perform controlled human exposure to freshly generated diesel exhaust following segmental allergen exposure in the lower airways of atopic human subjects.
Introduction
While viral and allergenic exposures have been the primary focus of research on asthma exacerbation, the evidence linking combustion-derived particulate matter (PM)1 to asthma symptoms and exacerbations is also considerable.2 Worldwide ambient PM exposures cause a loss of approximately 3% of global healthy life years.3 Diesel exhaust (DE) is a key source of ambient PM less than 2.5 µ in diameter (PM2.5),4 which penetrates deeply into the lung and has been strongly associated with acute deterioration of asthmatic lung function.5
Allergen challenge and segmental allergen challenge (SAC) are clinically important tools to examine the pathophysiology of immune-mediated disease.6 SAC has elucidated the role of eosinophils in response to allergen exposure.7 ,8 Combustion particles appear to act as carriers of allergens, thereby facilitating the entrance of allergens into the airways.9 Air pollutants may also exert adverse effects indirectly by acting on allergens. Allergen morphology and allergenicity appear modified following interactions with air pollutants.10 ,11 Accordingly, a model of combined exposure to air pollution and allergen is desirable.
Rodent models and in vitro studies using human-derived cells have shown that particles derived from DE augment allergenic effects on Th2 cytokines, eotaxin and eosinophil activities.12–14 DE-associated augmentation of select allergenic phenomena was shown in a human nasal model,15 ,16 and similar findings were noted with other particulate exposures.17 ,18 Still, DE's enhancement of allergenic effects has never been demonstrated in vivo in the distal human lung, a site of pathological importance in allergic disease, and the underlying details of this potential synergy are poorly understood.
Therefore, we hypothesised that a 2 h inhalation of DE at 300 µg PM2.5/m3 would augment the lower airway allergic inflammatory response to SAC in atopic subjects. Our primary outcomes were bronchoalveolar lavage (BAL) eosinophils and eosinophilic activation; secondary outcomes were markers of non-allergic inflammation (other BAL inflammatory cell counts) and adaptive and innate immune responses (dendritic and T-cell surface marker activation, specific IgE and cytokine levels). We predicted that our end points would increase in a specific ranked order, with the baseline (filtered air and saline (FAS)) condition followed by DE, allergen and DE plus allergen. Furthermore, we interrogated whether individuals with common polymorphisms of genes associated with metabolism of reactive oxidative species (glutathione-S-transferase M1 (GSTM1) and GSTT1) were particularly susceptible to the effects of co-exposure to inhaled DE and allergens, given the reported influence of these genes on human response to air pollution.19 ,20
Methods
Ethics
The ethical review boards of the University of British Columbia and the Vancouver Coastal Health Research Institute approved the study. Written informed consent was obtained from each participant. The study was registered at ClinicalTrials.gov (trial no. NCT01792232).
Subjects and exposure protocols
We recruited for the following inclusion criteria: age 19–49 years, non-smoking, able to provide informed consent by local advertising and referral/solicitation of clinic patients and skin prick-positive to one of our three study allergens: birch, Pacific grasses and house dust mite (Dermatophagoides pteronyssinus group 1). On primary screening we excluded those with any of the following: (1) pregnancy/breastfeeding, (2) use of inhaled corticosteroids, (3) regular use of bronchodilator medication (ie, use of bronchodilators more than three times per week), (4) unstable asthma symptoms, (5) any use of vitamins A, C, E or antioxidant supplements, (6) comorbid conditions judged by the investigators to increase risk of dropout or (7) work in an industrial setting or other setting of significant inhaled exposures. For detailed determination of study allergen and other screening procedures, see the online supplement. Consented subjects entered a crossover experiment using two conditions (DE at 300 µg PM2.5/m3 or filtered air (FA); table 1) each for 2 h, the order of which was randomised and counterbalanced. DE exposure generation used a previously described system that excludes potential contamination with lipopolysaccharide,21 except that in the present study we used a 2.5 kW constant load.
Supplemental material
Inhaled exposure characteristics
We provide a representative image of the PM generated by the 2.5 kW constant load (figure 1), in addition to table 1. The mean temperature was 24.2°C (SD 0.5°) and the mean relative humidity was 35.3% (SD 2.0). Notably, the protocol allows for effective blinding of both the subjects22 and those analysing all material and data. Figure 2 summarises the crossover design, including the SAC. Briefly, we used skin prick testing to identify sensitisation to a study allergen and then to choose the concentration for the segmental challenge of that allergen. The allergen and diluent control were instilled in right middle and lingular sub-segments and the identical sub-segments were entered 48 h later for sampling. This is detailed further in the online supplement. This effectively results in four exposure conditions per subject: FAS, diesel exhaust and saline (DES), filtered air and allergen (FAA) and DEA (diesel exhaust and allergen). The following assays were performed on the airway samples (FAS, DES, FAA, DEA) collected 48 h post exposure. Details of bronchial wash (BW) and BAL are provided in the online supplement.

Transmission electron microscopy (TEM) image of diesel exhaust particle collected from the Air Pollution Exposure Laboratory exposure booth. Diesel exhaust particles were collected from the engine at a load of 2.5 kW, corresponding to the regular exposure conditions. The particles were collected on a carbon-coated TEM grid.
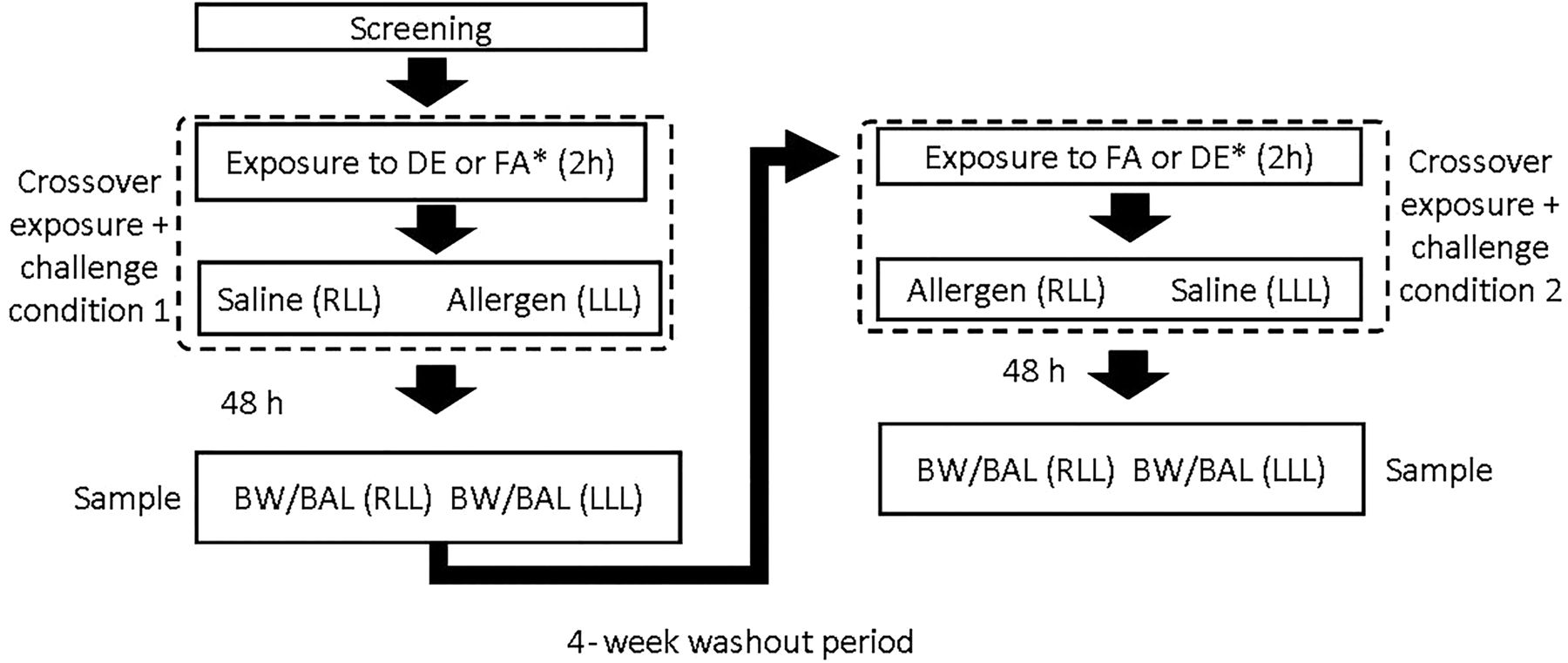
Outline of sequence of exposure and sampling. All subjects received DE and FA, separated by washout; subjects were randomised to received either DE or FA first. BAL, bronchoalveolar lavage; BW, bronchial wash; DE, diesel exhaust; FA, filtered air; RLL, right lower lobe; LLL, left lower lobe.
Cell differential (BW and BAL)
A total of 30 000 cells were spun down and stained with Harleco Hemacolor kit (EMD Millipore, Billerica, Massachusetts, USA) for each cytospin. Two observers counted at least 400 cells for every sample, and total cells (cells/mL of lavage fluid) and differential (percentage) cells were counted for macrophages, neutrophils, bronchial epithelial cells, eosinophils and lymphocytes. The counts were averaged and the interobserver variability was confirmed to be less than 10% for all samples.
Cytokines (BW and BAL)
The concentrations of interleukin (IL)-1Ra, IL-6, IL-8, interferon gamma-induced protein 10 (IP10), monocyte chemotactic protein (MCP)-1, major intrinsic protein (MIP)-1β, and RANTES were determined in both BW and BAL. IL-5, IL-13, IL-15, CCL-20, eosinophil cationic protein (ECP), granulocyte-colony stimulating factor (GCSF), 8-isoprostane, tumour necrosis factor-α and vascular endothelial growth factor were determined only in BAL due to smaller volumes in BW. They were assessed using the Luminex assays (Human Cytokine/Chemokine Magnetic Bead Panels I and II and the Human Th17 Magnetic Bead Panel, EMD Millipore Corporation, Billerica, Massachusetts, USA). Cytokine concentration units are given in picogram/millilitre or nanogram/millilitre (ECP). The limits of detection were as listed in the product literature.
T-cell subsets, eosinophil and dendritic cell characterisation and activation (BAL)
We assessed percentage T-cell subsets (cytotoxic, helper, regulatory), percentage eosinophils and percentage dendritic cells (DCs) and each of their cell surface phenotype activation (in terms of mean fluorescence intensity (MFI)) by flow cytometry using BD FACSCanto II. For all staining, cells were pelleted, resuspended in 50 μL phosphate-buffered saline and then incubated for 30 min. Specific markers and associated fluorophores are noted in Table E1 (see online supplement).
Allergen-specific IgE
The level of IgE specific to the allergen used in the segmental challenge for a given patient was assessed in BAL by the ImmunoCAP system, given its high sensitivity and its specificity similar to other methods.23
Genotyping
Individual status for GSTM1 and GSTT1 (present/null) was assessed due to the prominence of these genes in the air pollution literature, the high prevalence of variants in these genes in the general population and the known loss of function associated with the null genotype.19 ,20 Genotyping was by gel electrophoresis after multiplex PCR using DNA isolated from blood. β-globulin gene (primer pairs: forward 5′-CAA CTT CAT CCA CGT TCA CC-3′; reverse 5′-GAA GAG CCA AGG ACA GGT AC-3′) was used as an internal control. The primer pairs for each gene were as follows: GSTM1: 5′-GAACTCCCTGAAAAGCTAAAGC-3′, 5′-GTTGGGCTCAAATATACGGTGG-3′; GSTT1: 5′-TTCCTTACTGGTCCTCACATCTC-3′, 5′-TCA CCG GAT CAT GGC CAG CA-3′.
Statistical analysis
p Values less than 0.05 were considered significant. A mixed effects model was used to estimate all effects, and pertinent contrasts were used to test the hypotheses. Models included inhaled exposure (DE or FA), segmental exposure (allergen or saline), order (DE before FA or FA before DE) and gene variant status (GSTM1 present or null; GSTT1 present or null) as fixed effects, and subject identification number as a random effect. Inhaled exposure effect (‘DE’), segmental exposure effect (‘allergen’) and order effect were each assessed and are reported as such in the results. A mixed effects model was used to examine the potential interaction between DE and allergen exposure, statistically on the multiplicative level. We also performed analysis of ordinal contrasts (see online supplement) to assess our a priori hypothesis that our end points would be ranked in the following order: FAS<DES<FAA<DEA. Finally, we looked at GST interactions with exposure (as an ordered factor for those end points with a significant ordinal effect).
Results
Our study enrolled 11 women and 7 men, aged 20–46 years (table 2). Of these, six had baseline hyper-responsiveness to methacholine (PC20 ≤8 mg/mL); three of these six were doctor-diagnosed with asthma. Baseline FEV1 ranged from 66% to 143% predicted; baseline PC20 ranged from 0.23 to >16 mg/mL methacholine. One subject did not consent to genotyping; 10/17 were genotype null for GSTM1 and 8/17 were genotype null for GSTT1.
Demographics of 18 subjects enrolled in study
All 18 subjects completed all four bronchoscopies. However, analysis was not based on 72 (18×4) samples for each end point because of variable return from the washings; specifically, for a given subject, the ability to compare all four samples for all end points was limited by the return (of the four samples, whether from BW or BAL) with the lowest yield. Once that subject’'s lowest-volume return was fully used, then four-way comparisons for that subject were no longer possible. This (running out of sample for at least one of the four conditions) did occur more frequently for BW (as expected, given lower volume relative to BAL) such that the number of subjects with all four conditions for comparison was as follows, per end point: cytokines: BAL=18, BW=15; IgE: BAL=17, BW=17; cell counts: BAL=15, BW=11 and flow cytometry: BAL=16, not performed on BW.
No adverse clinical events were reported. However, as detailed below, DE generally augmented allergen-induced markers of both allergic and non-allergic inflammation, along with innate and adaptive immune responses, and null status for GSTT1 (but not GSTM1) further enhanced the increases in some of these end points.
Changes in number and proportion of airway leucocytes and macrophages
In BW, lymphocytes and eosinophils cell numbers decreased with DE, reflecting the trend of decreasing overall cell number with DE. Eosinophils (total and percentage) increased with allergen (table 3). In BAL, allergen increased total cells, neutrophils (total and percentage) and eosinophils (total and percentage) and decreased macrophages (percentage) and there was a significant ordinal effect on neutrophils, macrophages and eosinophils and total cells. Other changes were not significant.
Bronchial wash (BW) and bronchoalveolar lavage (BAL) cell counts
Changes in number, proportion and cell surface marker activation of airway dendritic cells, macrophages and T cells
DC, macrophage and T-cell quantification and phenotyping were performed on BAL cells (table 4). CD4 expression in helper T-cells, Foxp3 expression in regulatory T cells and cytotoxic and regulatory T-cell activity (MFI) increased with allergen. There was a positive ordinal effect on CD4 expression in helper T cells. CD86 expression of macrophages and DCs was suppressed by DE alone. Other changes (whether in terms of total number or percentage) were not significant.
Changes in T-lymphocyte subsets and activation of these subsets as well as eosinophil and dendritic cells by exposure condition, according to flow cytometry analysis of bronchoalveolar lavage
Change in airway cytokines and chemokines
Cytokine and chemokine measurements performed on BW and BAL were differentially influenced by exposure condition (table 5). BAL levels of IL-15 decreased with DE, while IL-1Ra, IL-8, IP10 and MCP-1 in BW increased with allergen. IL-8, chemokine (C-C motif) ligand 20 (CCL20), ECP, GCSF, IP10 and MCP-1 in BAL increased with allergen. There was a positive ordinal effect on IL-8, ECP and MCP-1 in BAL. Other changes were not significant.
Bronchial wash (BW) and bronchoalveolar lavage (BAL) cytokines
Change in allergen-specific IgE
The BAL allergen-specific IgE levels were below the limit of detection (0.35 kU/L) in 53 of 64 samples (from 16 subjects with sufficient lavage fluid in each of four exposure conditions), such that it was not feasible to perform an analysis based on average levels of specific IgE. Therefore, we instead considered the number of samples in each condition that was above the limit of detection and this was most frequently the case with DEA (5/16) versus the other conditions (FAS: 1/16; DES 2/16; FAA 3/13), but this trend was not significant (p=0.17).
Additive and multiplicative interactions
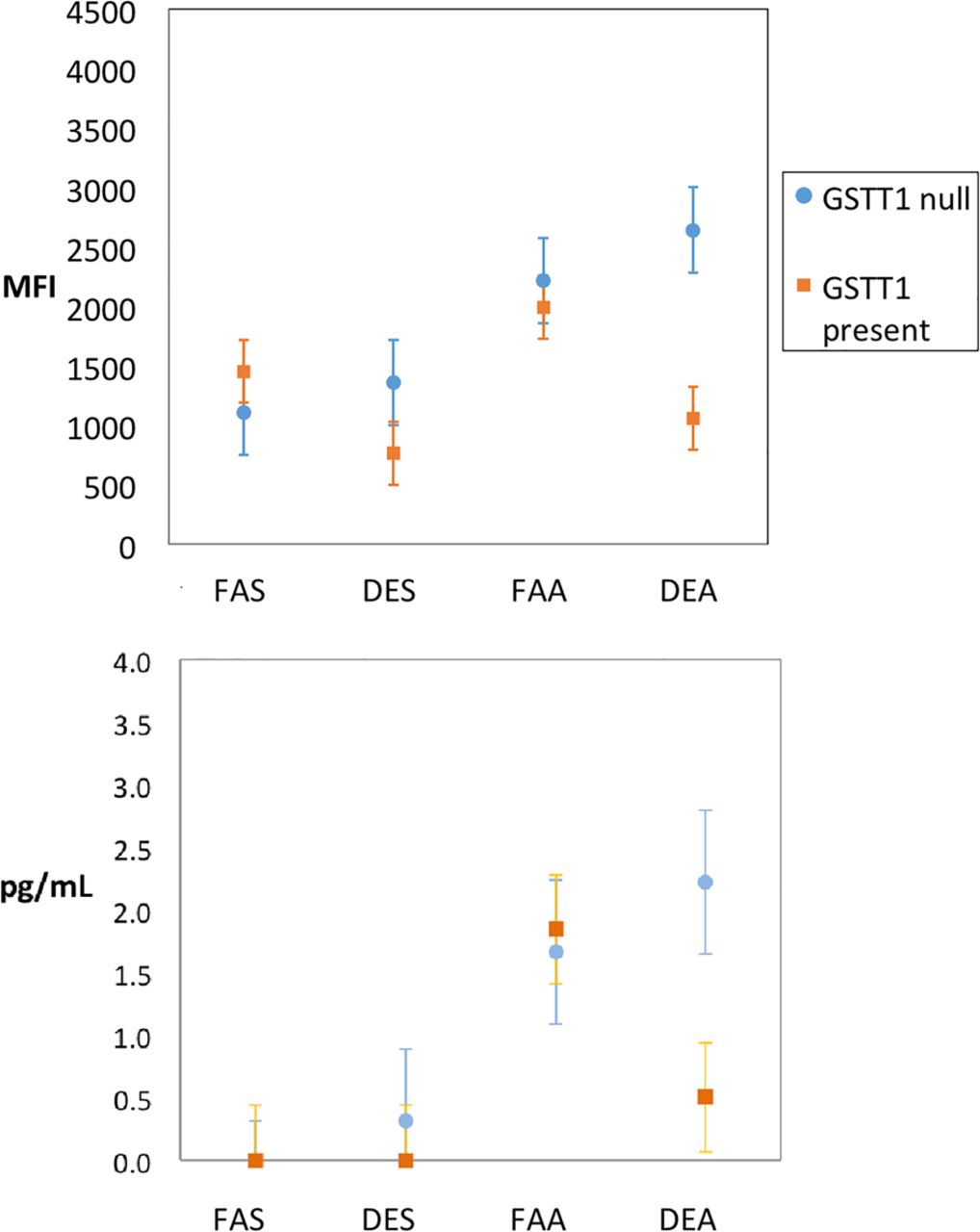
There were no multiplicative-level interactions between DE and allergen. The seven end points with significant ordinal effect (additive-level interaction) are represented graphically in figure 3. Two end points were significant in terms of interaction between exposure conditions and GSTT1, indicating that the effect was strongest in those with the GSTT1 null genotype for BAL IL-5 (interaction p=0.02) and activation of cytotoxic CD69+ T cells (interaction p=0.01; figure 4).

End points in bronchoalveolar lavage with positive linear ordinal effect, separated by exposure combinations as designated (filtered air and saline (FAS); diesel exhaust and saline (DES); filtered air and allergen (FAA); diesel exhaust and allergen (DEA)). Circles and squares represent means; whiskers are the upper and lower bounds of the 95% CI. IL, interleukin; MFI, mean fluorescent intensity.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of IL-5 (A) and cytotoxic T-cell activity (B) in bronchoalveolar lavage according to GSTT1 genotype and separated by exposure combinations as designated (filtered air and saline (FAS); diesel exhaust and saline (DES); filtered air and allergen (FAA); diesel exhaust and allergen (DEA)). Circles represent means; whiskers are the upper and lower bounds of the 95% CI. IL, interleukin; MFI, mean fluorescent intensity.
Discussion
The plausibility of synergisms among environmental pollutants and allergens to induce enhanced allergic responses in atopic individuals is generally accepted but specific human in vivo experimental validation of this concept is difficult to achieve and hence limited. Accordingly, we implemented a novel experimental exposure study (DE followed closely by allergen exposure) that tested the hypothesis that DE augments the effects of allergen in the lower airway. We measured a range of allergic inflammatory and immune-related end points in response to single and combined exposure to DE and allergen, and examined the role of GST genotypes in terms of potential to confer increased risk for DE-augmented responses.
Overall, we report that DE has an additive effect on several markers of allergic inflammation (eosinophils, IL-5, ECP) as well as non-allergic inflammation (neutrophils, IL-8, MCP-1). This is an important finding given that our experimental DE level is consistent with short-term real-life ambient exposures,24 ,25 in low/middle-income countries in Asia,26 ,27 and in occupational settings,28 ,29 though we recognise that these levels are uncommon in most ambient settings and at the upper end within the range of prior controlled human studies.21 That said, these findings differ from similar earlier studies of DE alone, where inflammatory markers like ECP and MCP-1 were not reported or reported markers (neutrophils, IL-8) were from the large airways and not from distal alveolar lung regions.30 This raises the possibility that the addition of allergen may variably affect different regions within the airway, either due to airway biology that varies regionally (with equal amounts of allergen) and/or due to variable regional dosing of allergen (perhaps disproportionately distal, due to gravity, in the context of SAC). We found no data in the literature or by expert consultation (personal communication with Dr Nizar Jarjour) for the lattermost speculation, but this would be an important point to explore in future study. Inhaled PM is known to deposit regionally primarily based on size (diameter), with the smallest particles deposited disproportionately in more distal regions, and this may be the reason why synergistic effects were generally more prominent in the BAL (tables 3 and 5).
An important detail in the current study is the requirement that participants be allergen-sensitised rather than asthmatic per se, as reflected by the inclusion criteria and supported by the observation that atopy alone is sufficient to elicit lower airway allergic inflammation on local allergen challenge.31 To probe if our observations tended to be greater in atopic asthmatic subjects relative to atopic non-asthmatics, we performed post hoc analysis of the effect of asthmatic status (only 6 of 18 subjects having baseline airway hyper-responsiveness and only three of these six having doctor-diagnosed asthma). We observed no significant effect of asthma status on our outcomes; this is likely due to being underpowered for an analysis focused specifically on asthma status, which was not the intention of our study. Accordingly, we neither expected nor observed any significant change in airway responsiveness given that the minority of our subjects had airway hyper-responsiveness at baseline, which we have already shown is the key predictor of a DE-attributable change.32 The focus on allergen-sensitised participants in our study allows demonstration that even in atopic non-asthmatics, inflammation may be induced in the lower airways without clinical symptoms of airway dysfunction. Although our study was not designed to directly address the issue of ‘atopic march’, a concept whereby atopy may develop into clinical asthma, our findings support this concept, albeit indirectly. Specifically, we speculate that a combined exposure creates an immunological milieu that is capable of enhancing inflammation relevant in asthma development and exacerbations. The impact of combination exposures is particularly pronounced in those atopic individuals without a normally functioning GSTT1.
The significant changes in some of the endpoints in this study merit further interrogation. The augmentation by DE on allergen-induced MCP-1 is relevant given that MCP-1 recruits antigen-presenting cells and T cells to sites of inflammation.33 Notably, T-helper cells as assessed by flow cytometry were significant in the ordinal analysis and consistent with the pattern for MCP-1 (figure 3), although activity in DCs was not increased (table 4).
Notably, those deficient in GSTT1 showed increased activation of cytotoxic T cells only in the context of DE plus allergen, suggesting that there are important exposure–exposure–gene interactions that may be missed without detailed probing. Similarly, the finding that IL-5 was increased significantly only in those deficient in GSTT1 is interesting in light of prior nasal data16 in which allergen-induced IL-5 was significantly enhanced by DE particles, but GSTT1 status was not examined. Given the literature on the importance of the GST family in responding to oxidative insults including air pollutants like ozone,34 our findings on GSTT1 are consistent in the context of air pollution;20 we cannot explain why GSTT1 had more influence than GSTM1, except to note that such inconsistency is common in the air pollution literature when various GST isoforms have been examined.
Considering the effect of single exposures, allergen had a more pronounced effect than DE on the end points examined; allergen independently increased airway neutrophils, eosinophils, IL-1Ra, IL-8, CCL20, ECP, GCSF, IP10, MCP-1 and CD4 expression in helper T cells. None of these changes are surprising, given known effects of allergen in the airway, and they validate our SAC model. It may be surprising that IL-5 did not significantly increase in response to allergen, but this appears to be due to large variability in response (although the absolute change (2.3–51.2 pg/mL) is very similar to that observed in a prior investigation).35 A unique finding, not previously reported in the context of human SAC models, is the allergen-induced increase in Foxp3 expression in regulatory T cells. Interestingly, DE did not have an independent effect on regulatory T cells. This contrasts with the study of Nadeau et al,36 where a cross-sectional analysis showed impaired T-regulatory cell function in those living in a region with higher ambient ozone and PM levels. The contrast likely has to do with differences in exposure frequency and/or duration, tissue specificity of exposure and sample collection, and the difficulty in controlling for allergen co-exposure in the observational setting, highlighting the need for more research in this area. Our findings of diesel-induced decrease in DC and macrophage activation is built on previous findings in a model that did not include allergen.37
Our blinded crossover study design has several strengths. Inhaled DE is an improvement over studies using instilled (rather than inhaled) particulate, often at unrealistic concentrations, or narrow and/or artificially generated fractions of the ambient pollutant mix.38 Bronchoscopy with SAC is novel in this context (never before applied to a human model of DE) and advantageous because it allows us to obtain, within the same bronchoscopy, samples reflecting exposure plus saline and exposure plus allergen. We had 50% more power than a similar study recently published using spark-generated ultrafine particles; this increased power may be an explanation to our finding effects in end points (such as IL-5 and IL-8) that were unchanged in that study.38 An additional, or alternative, explanation for our distinct findings may be the use of diluted and aged DE, a more realistic reflection of traffic-related air pollution, which includes nitrogen dioxide (itself known to enhance eosinophilia at concentrations similar to those we employed).39 We previously saw effects of combined exposure to allergen and combustion products on the chronic40 timescale but now have contributed to understanding more acute dynamics and thus appreciate the effects of common co-exposures in more detail. Our 48 h delay between exposure and airway sampling may be advantageous in two respects: (1) it provides insight into effects beyond the more typical 24 h sampling window; (2) it may decrease any potential sequence-related effect (as reported by others;38 our post hoc analysis revealed no such effect on the end points we evaluated).
Our study also has important limitations. The 48 h delay between exposure and lung sampling, while helpful for capturing changes related to adaptive immunity, may miss more immediate effects that peak earlier in the time course. However, the changes noted at 48 h are remarkable in their persistence at post-exposure time points later than those typically studied in such controlled experiments. Furthermore, the segmental allergen instillation, though methodologically efficient and enables the study of airway cells otherwise not accessible during a natural allergen-induced exacerbation, is still not entirely realistic; the segment receiving the challenge dose likely has a higher than physiological dose, while at the same time the whole lung is receiving a lower than physiological dose. We also recognise that since we excluded those with more severe disease, it remains unclear to what extent our findings would be similar in those with more significant clinical airways disease.
In summary, we have employed a novel protocol that could exploit several advantages in terms of co-exposure, sample recovery and timing. We used a chamber exposure to DE with SAC and bronchoscopy to assess the ability of DE inhalation to augment allergen-induced allergic inflammation in atopic individuals. We have demonstrated important links between allergic sensitisation, air pollution and increased susceptibility to DE. Unfortunately, the global emissions problem is not going away.41 Furthermore, global climate change is leading to increases in both PM and allergens,42 thus potentially compounding the adverse effects of dual exposure to these inhalants. With this step in understanding the complexity of the in vivo human reaction to multiple exposures, our study serves a critical public health objective, namely protecting vulnerable populations from air pollution. This report contributes evidence of susceptibility and further supports biological plausibility of epidemiological findings showing increased risk to atopic individuals.
Acknowledgments
The following individuals graciously offered valuable discussions at various stages of the project: Drs Michael Brauer, Sarah Charlesworth, Ramin Dastanpour, Mark FitzGerald, Stuart Turvey and Zhihong Zhang. The authors would also like to acknowledge the subjects who participated in the research study for their commitment to medical research.
References
Footnotes
Contributors CC conceived of and recruited subjects for the study. CC, NA, AB, MP, TS and JH refined the details of the methodological approach. MP and SWW performed the analyses. CC primarily wrote the manuscript. All authors read, edited and approved the final manuscript.
Funding This study was supported by the Canadian Institutes for Health Research (MOP 123319), WorkSafe BC (RG2011-OG07) and AllerGen NCE (GxE4).
Competing interests None declared.
Ethics approval University of British Columbia Clinical Research Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction