Article Text

Abstract
Background Recently, several proteins of the extracellular matrix have been characterised as active contributors to allergic airway disease. Microfibrillar-associated protein 4 (MFAP4) is an extracellular matrix protein abundant in the lung, whose biological functions remain poorly understood. In the current study we investigated the role of MFAP4 in experimental allergic asthma.
Methods MFAP4-deficient mice were subjected to alum/ovalbumin and house dust mite induced models of allergic airway disease. In addition, human healthy and asthmatic primary bronchial smooth muscle cell cultures were used to evaluate MFAP4-dependent airway smooth muscle responses.
Results MFAP4 deficiency attenuated classical hallmarks of asthma, such as eosinophilic inflammation, eotaxin production, airway remodelling and hyperresponsiveness. In wild-type mice, serum MFAP4 was increased after disease development and correlated with local eotaxin levels. MFAP4 was expressed in human bronchial smooth muscle cells and its expression was upregulated in asthmatic cells. Regarding the underlying mechanism, we showed that MFAP4 interacted with integrin αvβ5 and promoted asthmatic bronchial smooth muscle cell proliferation and CCL11 release dependent on phosphatidyloinositol-3-kinase but not extracellular signal-regulated kinase pathway.
Conclusions MFAP4 promoted the development of asthmatic airway disease in vivo and pro-asthmatic functions of bronchial smooth muscle cells in vitro. Collectively, our results identify MFAP4 as a novel contributor to experimental asthma, acting through modulation of airway smooth muscle cells.
- Asthma Mechanisms
Statistics from Altmetric.com
Key messages
What is the key question?
Here we investigated the role of MFAP4 in experimental asthma.
What is the bottom line?
This study shows for the first time that MFAP4 is involved in the pathophysiology of asthmatic airway disease as a modulator of airway smooth muscle biology.
Why read on?
The study identifies a novel pathway involved in bronchial smooth muscle cell modulation in asthma pathogenesis.
Introduction
Asthma is a chronic airway disease characterised by eosinophilic inflammation, airway hyperresponsiveness (AHR) and airway remodelling, and the interplay between these three components is still incompletely understood. Common features of asthmatic remodelling include changes in the composition of the extracellular matrix (ECM) as well as airway smooth muscle (ASM) hyperplasia and hypertrophy.1 ASM interactions with the surrounding ECM have been demonstrated to be involved in ASM thickening. ECM components, and matrix derived from subjects with asthma, have been shown to increase healthy ASM proliferation.2 ,3 Thus, changes in ECM composition and quantity may influence the function of neighbouring ASM cells.
The ASM–ECM interaction is mediated mainly through specific integrin receptors. Function-blocking studies have shown that β1 and αvβ3 integrins are responsible for fibronectin-mediated ASM proliferation and IL-1β-dependent cytokine production.2 ,4 Moreover, an RGD-containing blocking peptide has been shown to attenuate ASM remodelling in a guinea pig asthma model, underlining the importance of integrin signalling in asthma in vivo.5 The integrin-ligand interaction activates focal adhesion kinase (FAK) and further signal transduction, such as phosphatidyloinositol-3-kinase (PI3K) and extracellular signal-regulated kinase (ERK) pathways.6
Microfibrillar-associated protein 4 (MFAP4) is an ECM protein of the fibrinogen-related protein superfamily highly expressed in elastin-rich tissues, such as heart and lung.7 Due to elastin-binding properties, MFAP4 was suggested to contribute to elastogenesis and maintenance of the proper structure of elastic fibres.8 ,9 Importantly, MFAP4 contains an RGD sequence, suggesting that it can signal through integrin receptors, and we have shown that MFAP4 promotes vascular smooth muscle cell (SMC) proliferation and migration through integrins αvβ3 and αvβ5in vitro and in vivo (manuscript under revision). MFAP4 has also been suggested as a potential biomarker and, together with another ECM protein fibulin-5, as a contributor to abnormal tissue repair in COPD.10 ,11 Furthermore, we have recently shown that MFAP4-deficient mice exhibit mild age-dependent emphysema-like airspace enlargement.12
In the present study, we investigated the potential role of MFAP4 in the development of allergic asthma using two distinct murine models. Furthermore, we examined if effects of MFAP4 could be explained by ASM phenotypic modulation using in vitro culture of human bronchial SMCs (BSMCs).
Methods
Additional details on the methods are provided in the online supplementary material.
Mice
Female 8–12-week-old Mfap4−/−(knockout, KO) mice, generated previously (manuscript under revision), and littermate Mfap4+/+ (wild-type, WT) mice, were bred into the BALB/c background for more than 10 generations. The animals had access to pelleted chow and water ad libitum. The Danish National Animal Ethics Committee approved all animal experiments (permit 2012-15-2934-00354).
Allergic asthma models
Mice were sensitised intraperitoneally with 200 μg ovalbumin (OVA; grade VI) and 2 mg alum in 200 μL phosphate-buffered saline (PBS) on days 0 and 7. On days 14–16, mice were challenged intranasally with 20 μg OVA in 50 μL PBS under light isoflurane anaesthesia. Alum-sensitised, PBS-challenged mice were used as controls.
Alternatively, mice were challenged intranasally with 25 μg house dust mite extract total protein (HDM, endotoxin content 51.5 EU/mg protein) 5 days/week for 7 weeks as described previously.13 PBS-treated mice were used as controls.
Twenty-four hours after the last exposure mice were anaesthetised and subjected to AHR measurements. Briefly, mice were tracheostomised and connected to the ventilator (Flexivent, SCIREQ) and lung function parameters were measured in the steady state and after exposure to nebulised methacholine (MCh). Immediately afterwards mice were sacrificed and blood, bronchoalveolar lavage (BAL) fluid and lungs were collected. Serum IgE levels were determined by ELISA. Cytokine levels were quantified in BAL and lung homogenates. Hydroxyproline content was measured in lung homogenates.
Lung histology
Formalin-fixed, paraffin-embedded lung tissues were cut in 4 μm-thick slides and stained with H&E, periodic acid-Schiff (PAS), Picrosirius Red (PSR), or immunostained against α-smooth muscle actin (α-SMA) or MFAP4. The degree of inflammation, mucus production, fibrosis and ASM deposition was quantified by morphometric analysis.
Human subjects
Seven patients with severe persistent asthma and eight subjects without asthma were prospectively recruited from the CHU Bordeaux Teaching Hospital, Bordeaux, France, according to the Global Initiative for Asthma criteria.14 All subjects gave their written informed consent to participate in the study, after the nature of the procedure had been fully explained. The study followed recommendations outlined in the Helsinki declaration and received approval from the local ethics committee. Bronchial specimens from all subjects were obtained by either fiberoptic bronchoscopy or lobectomy, as previously described.15
Cell culture
Primary human BSMCs derived from healthy or asthmatic donors (Lonza) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50 U/mL penicillin, 50 μg/mL streptomycin and 2 mM L-glutamine. Cells between passages 5 and 10 were used in all experiments.
BSMCs were further isolated from bronchial specimens as described previously.15 Patient characteristics are presented in online supplementary table S1. Cells were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, 0.25 mg/mL amphotericin B, 2 mM L-glutamine, 1 mM sodium pyruvate and 1% non-essential amino acid mixture. Confluent cells were collected after 6–8 weeks. Cell purity and phenotype were validated by α-SMA and calponin positivity. Cells between passages 2 and 5 were used.
Real-time PCR
Relative expression of target genes was measured using Taqman Gene Expression Assays.
Detection of soluble MFAP4
MFAP4 levels in animal samples and BSMC lysates (prepared in PBS) were measured by AlphaLISA as described previously7 and shown as U/mL. 1 U/mL corresponds to 38 ng/mL in human serum.
Adhesion assays
Cell adhesion to ECM coating with or without pretreatment with either RGD and DGR-containing peptides or anti-integrin antibodies was measured using Vybrant Cell Adhesion Assay Kit.
Proliferation assay
Serum-starved BSMCs were seeded in serum-free DMEM on ECM-coated plates and incubated for 48 h. Cell proliferation was assessed using bromodeoxyuridine (BrdU) incorporation assay.
In some experiments, cells were seeded in serum-free DMEM for 4 h to allow attachment. Subsequently, anti-integrin antibodies or inhibitors of PI3K and mitogen-activated protein kinase (MEK) (LY294002 and PD98059, respectively) were added for the rest of the incubation period. Isotype control antibody and DMSO were used as controls for anti-integrin antibodies and inhibitors, respectively.
Western blotting
Serum-starved cells were incubated for various time points in serum-free DMEM and lysed in RIPA buffer with protease and phosphatase inhibitors. Equal amounts of total protein samples were separated by SDS-PAGE and blotted with antibodies against GAPDH, integrin αv, integrin β5, pFAK (T397) or FAK.
Statistics
Statistical analysis was performed in Prism 5 software (GraphPad, La Jolla, California, USA). Data are presented as means+SEM unless stated otherwise. Normality of the data was checked using D'Agostino-Pearson test. Parametric results were analysed using Student's t test or either one-way or two-way analysis of variance (ANOVA) followed by Bonferroni's multiple comparisons test. Non-parametric results were analysed using Mann–Whitney test. Correlations were analysed using Pearson's test. A value of p<0.05 was considered significant.
Results
Circulating MFAP4 levels are increased in allergic asthma
In this study we used two models of experimental asthma: an OVA-induced sensitisation and challenge model and a model of prolonged exposure to a clinically relevant allergen, HDM. Histological analysis revealed MFAP4 localisation within the basement membrane, in close proximity to airway epithelial cells and SMCs (figure 1A). MFAP4 mRNA expression did not change after OVA or HDM treatment (figure 1B). However, upon challenge the concentration of soluble MFAP4 was significantly increased in serum of WT mice (figure 1C). Moreover, we found that MFAP4 levels were raised in BAL of HDM-treated WT mice (figure 1D), with a corresponding decrease of MFAP4 content in lung homogenate (figure 1E).

Microfibrillar-associated protein 4 (MFAP4) is increased in bronchoalveolar lavage (BAL) and serum of allergic mice. (A) Pulmonary localisation of MFAP4 in ovalbumin (OVA) and house dust mite (HDM) treated mice. MFAP4 staining is shown with arrows. (B) MFAP4 mRNA expression level in the lung after OVA and HDM exposure. (C and D) MFAP4 levels are changed in asthma. MFAP4 concentrations were measured after OVA and HDM exposure in (C) serum, (D) BAL and (E) lung homogenate. Scale bar, 50 μm. n=2–14. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, calculated by Student's t test or Mann–Whitney test. PBS, phosphate-buffered saline.
MFAP4 deficiency attenuates allergy-induced airway eosinophilia and production of eotaxins
In both OVA and HDM models, MFAP4 KO mice showed decreased total cell and eosinophil numbers in BAL compared with WT littermates (figure 2A). The same pattern was observed in the lung parenchyma of OVA-treated mice, where WT animals exhibited more severe inflammation than their MFAP4-deficient counterparts (figure 2B, C). A trend towards reduced inflammation in the parenchyma was also observed in HDM-challenged MFAP4 KO mice (figure 2C). Moreover, we found a significant positive correlation between BAL eosinophil number and MFAP4 serum concentration in WT mice (figure 2D, E).

Microfibrillar-associated protein 4 (MFAP4) deficiency attenuates eosinophilic inflammation. (A) Both total cell count and eosinophil numbers in bronchoalveolar lavage (BAL) are lowered in MFAP4-deficient mice compared with wild-type (WT) littermates after ovalbumin (OVA) (left panel) or house dust mite (HDM) (right panel) treatment. (B) Representative pictures of H&E-stained lungs from OVA-treated (left panel) and HDM-treated (right panel) mice. (C) The degree of lung infiltration is lowered in MFAP4-deficient mice. Parenchymal inflammation was quantified in OVA-treated and HDM-treated animals. (D and E) Eosinophil counts in BAL correlated positively with MFAP4 concentration in serum of WT mice from (D) OVA and (E) HDM models. Scale bar, 100 μm. n=6 (OVA model), 10–11 (phosphate-buffered saline, PBS groups, HDM model), 14 (WT HDM), 19 (knockout, KO HDM). *p<0.05, **p<0.01, ***p<0.001, ns=not significant, calculated by one-way analysis of variance.
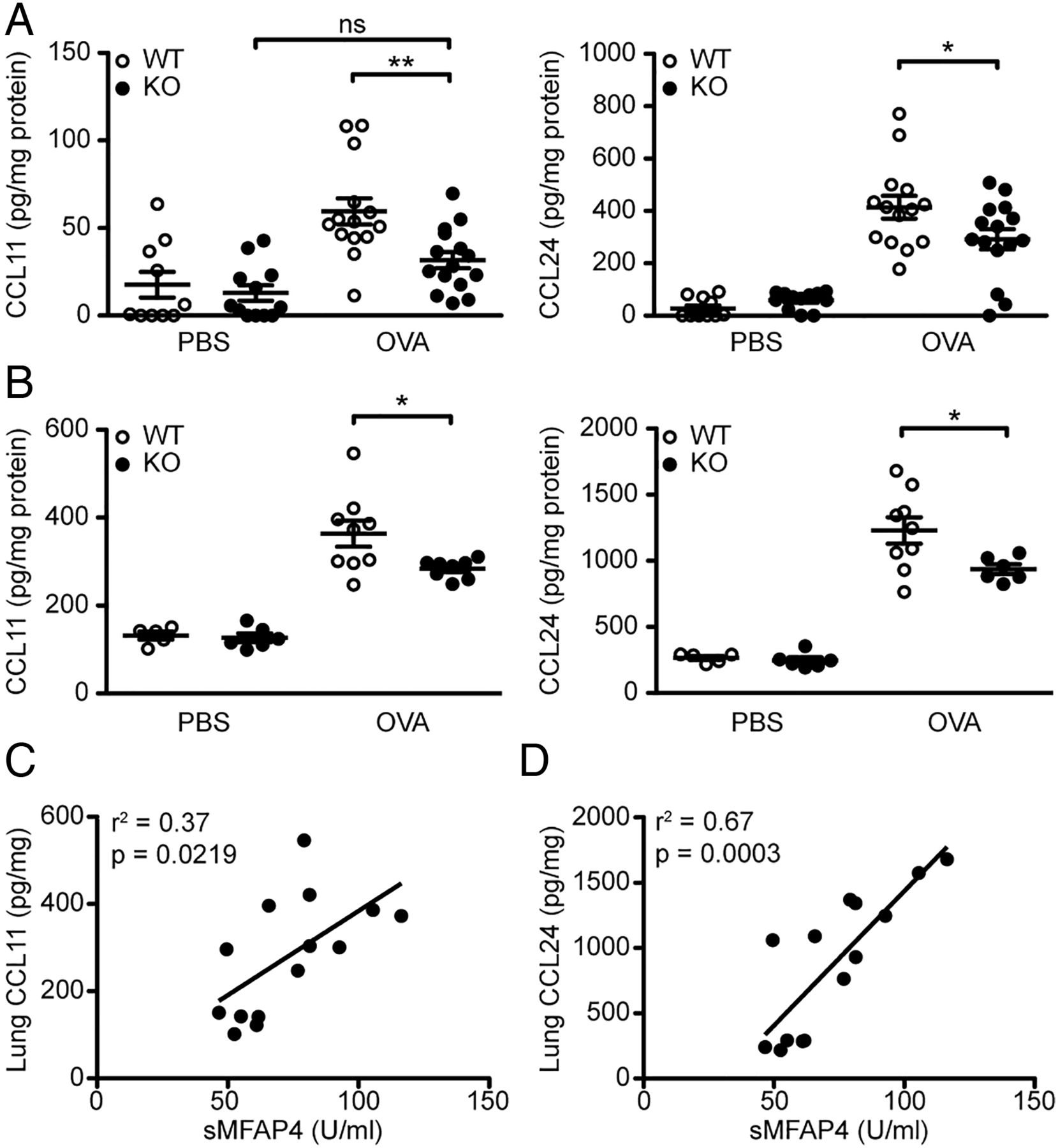
We then analysed the levels of CCL11 (eotaxin-1) and CCL24 (eotaxin-2), the chemokines essential for eosinophil recruitment. We found that OVA-treated MFAP4 KO mice had reduced amounts of CCL11 and CCL24 in BAL as well as in lung homogenates (figure 3A, B). Furthermore, the levels of lung CCL11 and CCL24 correlated with serum MFAP4 concentrations (figure 3C, D).

Microfibrillar-associated protein 4 (MFAP4) influences production of eotaxins. (A and B) Eotaxin-1 (CCL11) and eotaxin-2 (CCL24) were measured in (A) bronchoalveolar lavage (BAL) and (B) lung homogenate of ovalbumin (OVA)-treated mice. (C and D) Lung levels of both chemokines correlated positively with MFAP4 levels in serum of phosphate-buffered saline (PBS) and OVA treated wild-type (WT) mice. n=5–15. *p<0.05, **p<0.01, ns=not significant, calculated by one-way analysis of variance. KO, knockout.
MFAP4 influences IL-13 production
Th2 cytokine production was analysed in BAL and lung homogenates of OVA-treated mice. The levels of IL-4 and IL-5 did not differ between challenged WT and KO animals (see online supplementary figure S1A, B). However, we observed reduced production of IL-13 in BAL and lung homogenates of MFAP4-deficient mice (see online supplementary figure S1C). Serum OVA-specific IgE levels remained unchanged between treated groups (see online supplementary figure S2). Thus, in the used models MFAP4 did not appear to play an essential role in regulating Th2 responses but promoted IL-13 production.
Lack of MFAP4 attenuates goblet cell metaplasia
To determine whether mucus production was affected by MFAP4, lung sections were stained with PAS to evaluate the number of mucus-producing goblet cells (figure 4A). MFAP4 deficiency in OVA-treated mice resulted in a significant decrease in goblet cell number (figure 4B). We confirmed our observations in the chronic HDM model, where MFAP4 deficiency also resulted in downregulation of mucus production relative to WT mice, although to a smaller extent (figure 4C).
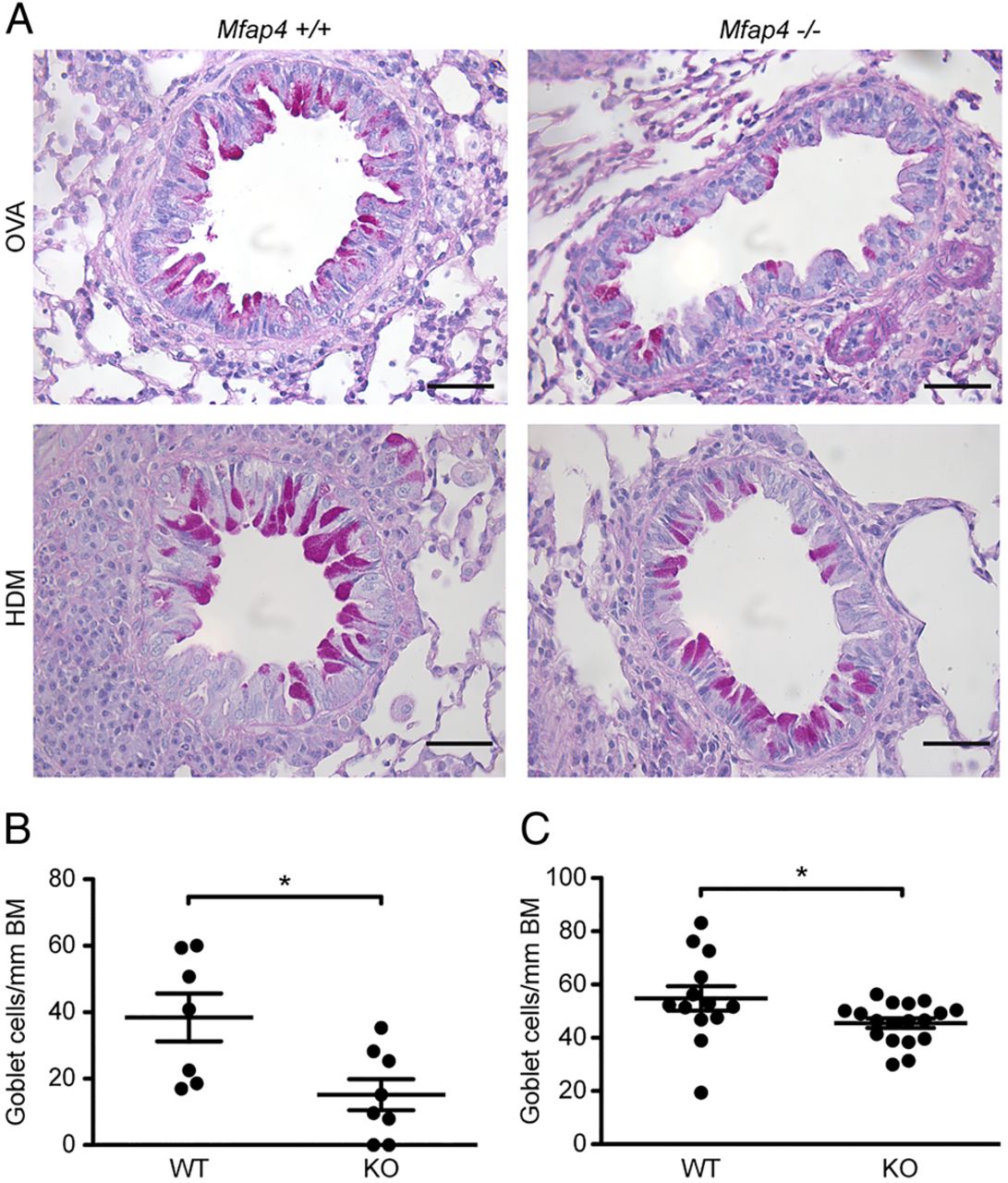
Microfibrillar-associated protein 4 (MFAP4) deficiency attenuates goblet cell metaplasia. (A) Representative pictures of periodic acid-Schiff (PAS)-stained lungs. (B and C) The degree of metaplasia was quantified in (B) ovalbumin (OVA)-treated and (C) house dust mite (HDM)-treated mice as the number of goblet cells normalised per length of the basement membrane (BM). Scale bar, 50 μm. n=7–19. *p<0.05, calculated by Student's t test. KO, knockout; WT, wild type.
MFAP4-deficient mice are partially protected from ASM deposition and fibrosis
To investigate the influence of MFAP4 on airway remodelling, we performed morphometric analysis of ASM deposition and fibrotic changes (figure 5A, B). The increase in peribronchial smooth muscle layer thickness was absent or significantly reduced in MFAP4 KO mice (figure 5C). Moreover, HDM-treated MFAP4-deficient mice had significantly lowered total lung collagen content (figure 5D). Accordingly, the collagen-specific PSR-stained area was significantly diminished in treated MFAP4-deficient mice (figure 5E). OVA treatment did not influence hydroxyproline content or peribronchial collagen deposition (data not shown), suggesting that this acute challenge protocol has not induced significant airway remodelling other than ASM hyperplasia.

Microfibrillar-associated protein 4 (MFAP4) promotes bronchial smooth muscle cell remodelling and fibrosis. (A and B) Representative pictures of (A) α-smooth muscle actin (α-SMA)-stained and (B) Picrosirius Red (PSR)–stained lungs. (C) Smooth muscle deposition was quantified in ovalbumin (OVA)-treated and house dust mite (HDM)-treated mice as the thickness of peribronchial α-SMA-stained layer. (D–E) Both (D) total collagen lung content and (E) peribronchial collagen-stained area are lowered in MFAP4-deficient mice. ASM, airway smooth muscle. BM, basement membrane. Scale bar, 50 μm (A), 100 μm (B). n=7–19. **p<0.01, ****p<0.0001, calculated by one-way analysis of variance. ASM, airway smooth muscle; KO, knockout; PBS, phosphate-buffered saline; WT, wild type.
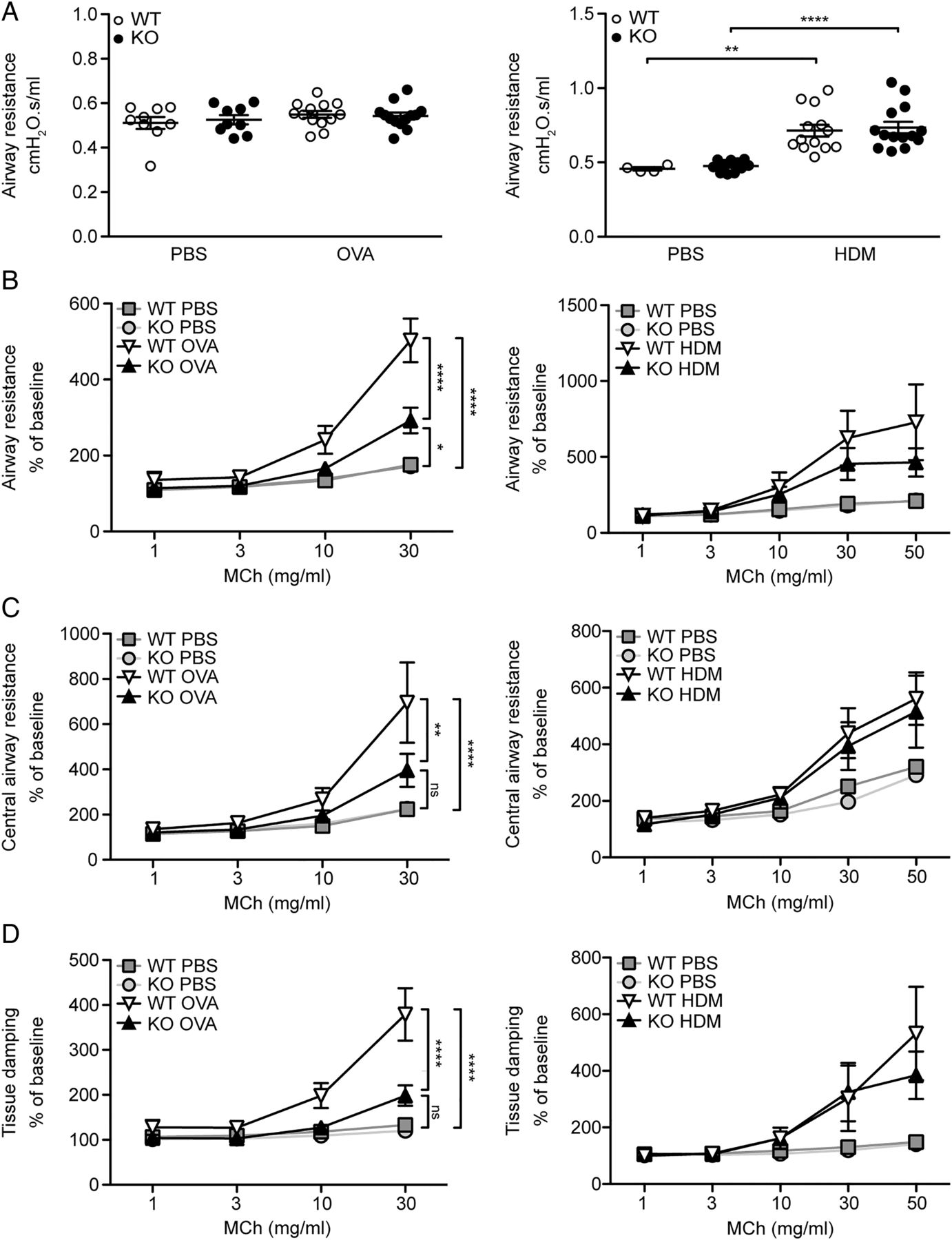
MFAP4 deficiency is protective against AHR
To specify the role of MFAP4 in AHR, we performed lung function analysis after MCh challenge. OVA exposure did not influence baseline resistance values, while chronic HDM treatment significantly increased baseline resistance independent of MFAP4 genotype (figure 6A). However, OVA-treated MFAP4-deficient mice demonstrated significantly lowered MCh-induced increase in pulmonary resistance than their WT littermates (figure 6B). The same tendency was observed in chronically treated animals (figure 6B). We then analysed data obtained from the so-called constant phase model, which divides resistance into central airway resistance of the conducting airways and tissue damping, a measure of resistance of lung parenchyma. We found that mainly changes in the parenchymal resistance are responsible for the overall attenuation of AHR in MFAP4-deficient mice (figure 6C, D).
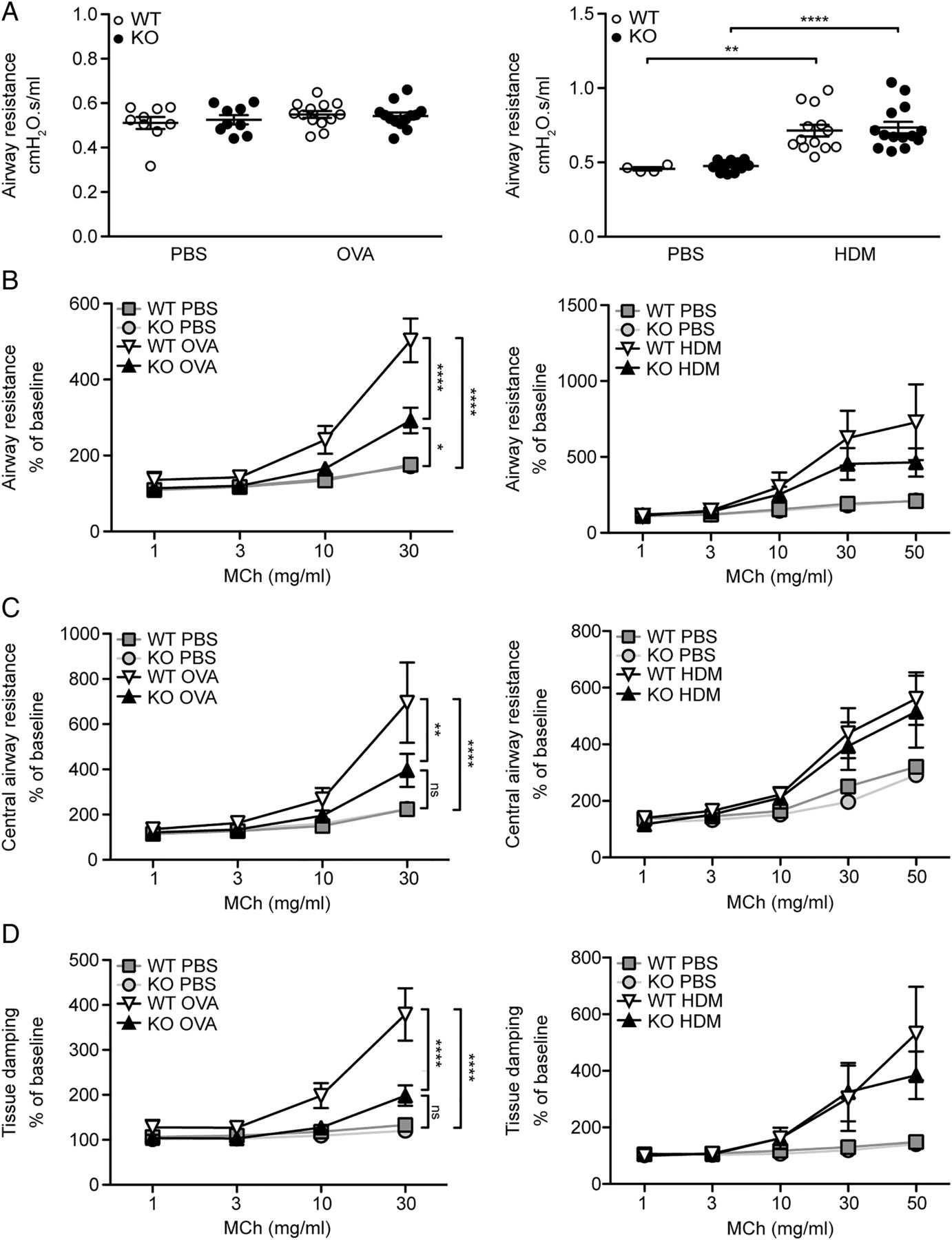
Microfibrillar-associated protein 4 (MFAP4) deficiency reduces airway hyperresponsiveness in ovalbumin (OVA)-treated but not house dust mite (HDM)-treated mice. (A) Basal airway resistance after OVA (left) or HDM (right) exposure. (B–D) Airway reactivity to increased doses of methacholine (MCh), measured as increase in (B) resistance, (C) central airway resistance and (D) tissue damping was strongly attenuated in OVA-treated MFAP4-deficient mice (left) but only slightly lowered in HDM-treated MFAP4-deficient mice (right). n=5–8 (OVA model), 5–13 (HDM model). *p<0.05, **p<0.01, ****p<0.0001, ns=not significant, calculated by two-way analysis of variance. KO, knockout; PBS, phosphate-buffered saline; WT, wild type.
MFAP4 is upregulated in asthmatic BSMCs
To further define the mechanism by which MFAP4 contributes to asthmatic airway disease, we studied the effects of MFAP4 on healthy or asthmatic human BSMCs in vitro. Using commercially available primary cells, we observed that asthmatic BSMCs produce increased levels of MFAP4 relative to BSMCs derived from the non-asthmatic donor, on mRNA (figure 7A) and protein level (figure 7B). We confirmed our initial observations on BSMCs isolated from a group of healthy subjects or patients with asthma, where we observed that mRNA and protein MFAP4 levels were clearly increased in the asthmatic group (figure 7C, D). The difference did not reach statistical significance, possibly due to large variation in the asthmatic group.
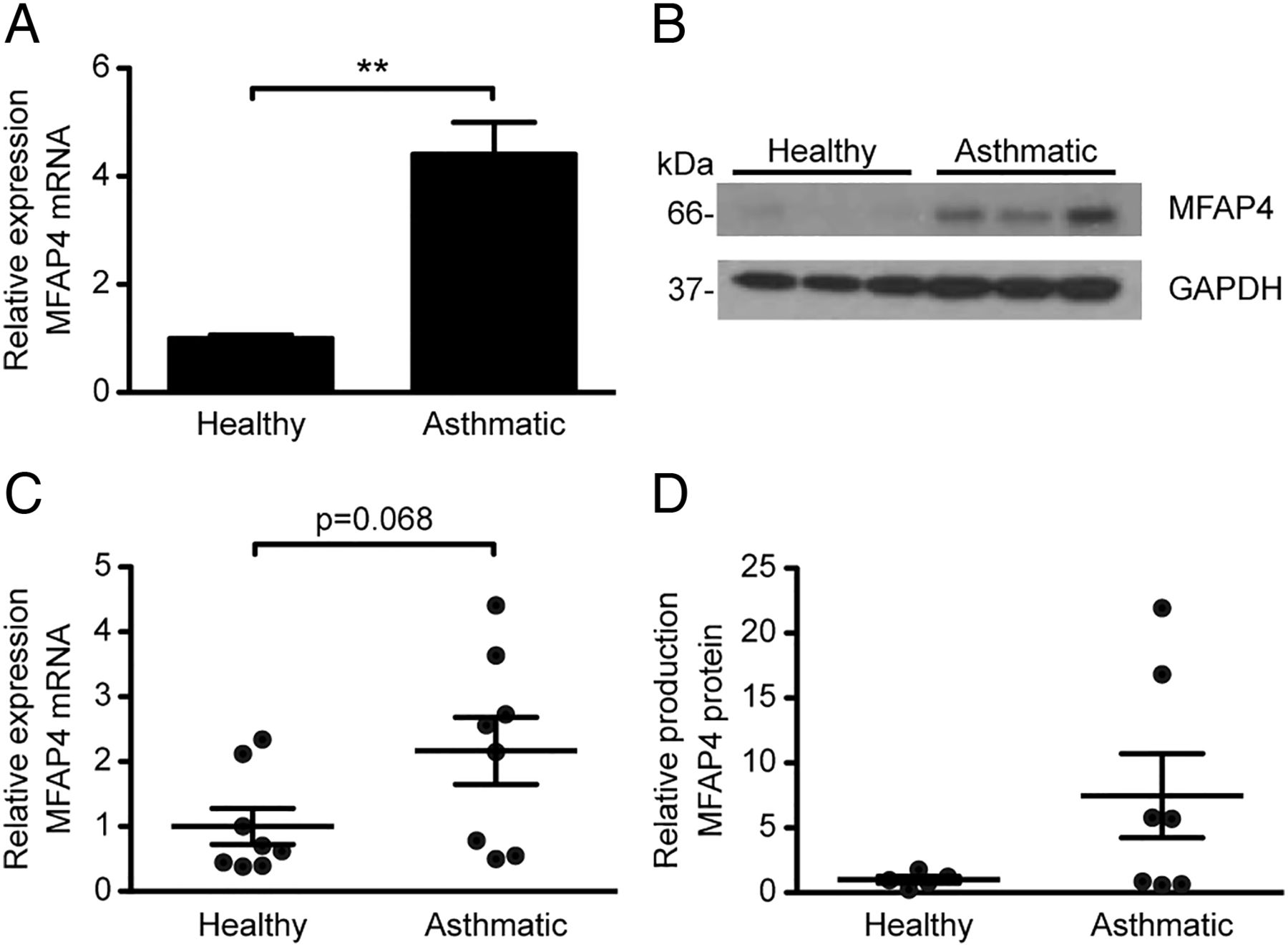
Microfibrillar-associated protein 4 (MFAP4) is upregulated in asthmatic bronchial smooth muscle cells (BSMCs). MFAP4 mRNA expression (A and C) and protein production (B and D) is increased in commercially available asthmatic BSMCs (A and B) as well as BSMCs isolated from patients with asthma (C and D) relative to healthy BSMCs. Data are means+SEM (A) or representative (B) of three independent experiments. n=5–8 (C and D). **p<0.01, calculated by Student's t test.
MFAP4 binds to BSMCs via integrin αvβ5 and activates FAK
We analysed expression of various integrin receptors on BSMC surface. We detected high levels of integrins β1 and αvβ5 and considerably lower levels of integrins β3 and αvβ3, whereas β6 and β8 subunits were undetected (see online supplementary figure S3). We then investigated the capacity and integrin dependence of MFAP4 to mediate BSMC adhesion. MFAP4 promoted adhesion of BSMCs in a dose-dependent manner, similarly in healthy and asthmatic cells (figure 8A) and to the level of the positive control fibronectin (data not shown). MFAP4-dependent adhesion could be inhibited with a soluble RGD peptide but not with a control DGR peptide, suggesting that MFAP4 binds RGD-dependent integrins on BSMC surface (figure 8B, C). Moreover, anti-αv and anti-αvβ5 antibodies prohibited BSMC adhesion, showing that αvβ5 is the main MFAP4-binding partner on BSMCs (figure 8D). MFAP4 stimulation did not influence protein abundance of either integrin subunit (data not shown).
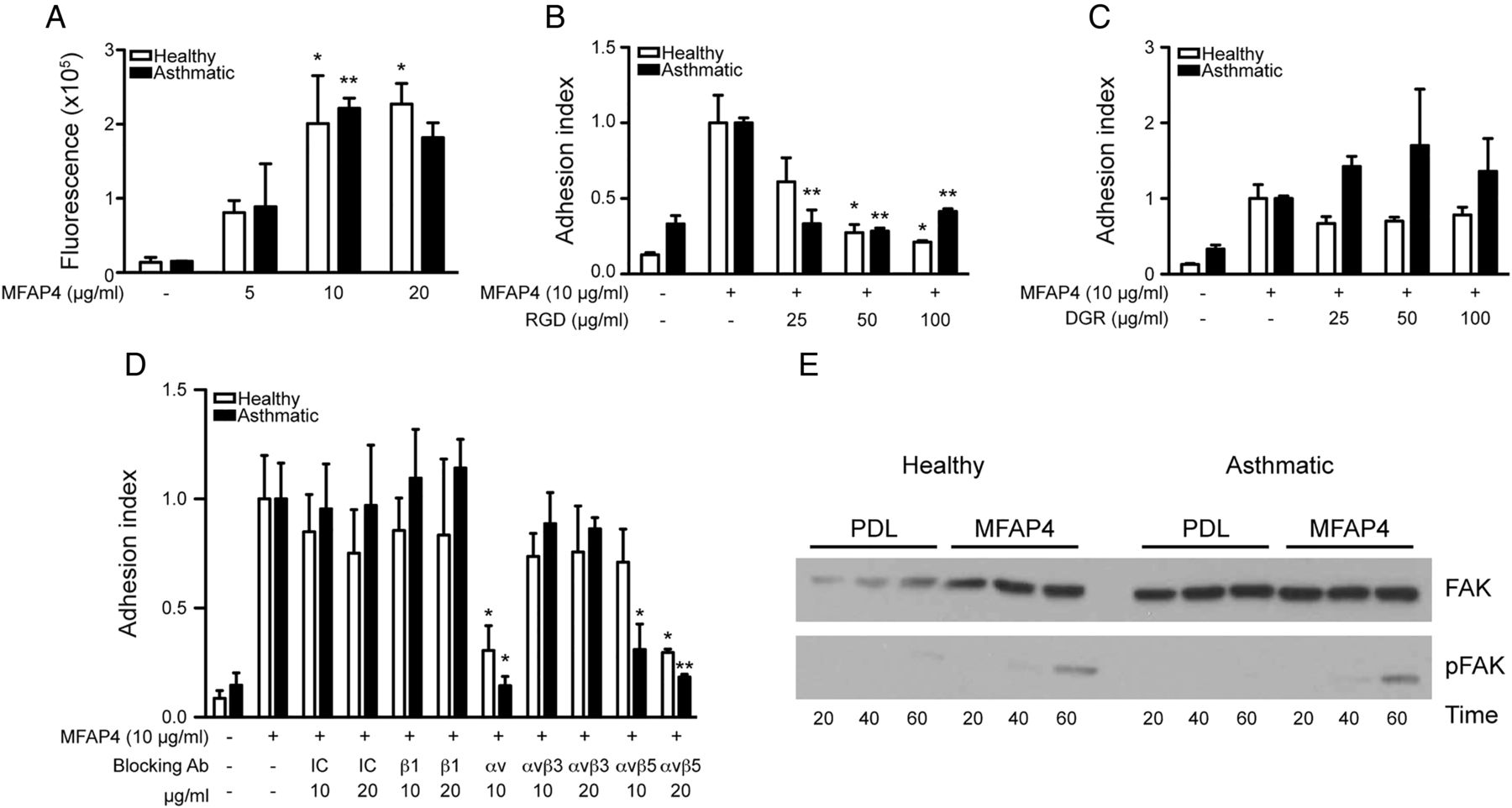
Microfibrillar-associated protein 4 (MFAP4) promotes bronchial smooth muscle cell (BSMC) attachment through integrin αvβ5 and focal adhesion kinase (FAK) activation. (A) MFAP4 promotes BSMC adhesion in a dose-dependent manner. (B and C) MFAP4-dependent adhesion can be inhibited by (B) RGD blocking peptide but not (C) DGR control peptide. (D) BSMC adhesion to MFAP4 is inhibited by integrin αvβ5-blocking antibodies. (E) Western blot analysis shows FAK phosphorylation after seeding cells on MFAP4. Data are means+SEM (A–D) or representative (E) of at least three independent experiments. *p<0.05, **p<0.01 compared with relevant uncoated (A), MFAP4-coated (B and C) or IC control (D), calculated by one-way analysis of variance. IC, isotype control; PDL, poly-D-lysine.
To verify if MFAP4 can promote initiation of cellular signalling, we checked time-dependent activation of FAK. Seeding healthy and asthmatic BSMCs on MFAP4 resulted in an increased time-dependent FAK phosphorylation relative to control cells seeded on poly-D-lysine (figure 8E).
MFAP4 promotes BSMC proliferation and CCL11 expression: role of αvβ5, PI3K and ERK1/2
We explored the influence of MFAP4 on the proliferative capacity of BSMCs. MFAP4 significantly increased BSMC proliferation in asthmatic BSMCs, while proliferation of healthy BSMCs was only slightly affected (figure 9A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Microfibrillar-associated protein 4 (MFAP4) promotes asthmatic bronchial smooth muscle cell (BSMC) proliferation and CCL11 expression through integrin αvβ5 and PI3K. (A) MFAP4 enhances asthmatic BSMC proliferation. (B) MFAP4-dependent asthmatic BSMC proliferation can be inhibited by integrin αvβ5-blocking antibody. (C and D) MFAP4-dependent asthmatic BSMC proliferation is blocked by PI3K inhibitor (C) but not MEK inhibitor (D). (E) MFAP4 enhances CCL11 mRNA expression in asthmatic BSMCs. Asthmatic cells seeded on MFAP4 (MFAP4, right) express higher levels of CCL11 than cells seeded on vehicle coating (control, left). (F and G) MFAP4-dependent CCL11 upregulation is inhibited by PI3K inhibitor (F) but not MEK inhibitor (G). DMSO was used as a vehicle control for kinase inhibitors. Data are means+SEM of at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001 compared with relevant coating control, calculated by one-way analysis of variance. IC, isotype control; LY, LY294002; OD, optical density; PD, PD98059; PDL, poly-D-lysine.
To further define signalling pathways involved in MFAP4-dependent BSMC proliferation, we cultured asthmatic BSMCs in the presence of anti-integrin antibodies or specific inhibitors of PI3K and MEK (MEK is an immediate upstream kinase crucial for ERK1/2 activation). We found that MFAP4-mediated proliferation was inhibited by anti-integrin αvβ5 blocking antibody (figure 9B). Moreover, MFAP4-dependent proliferation was inhibited dose-dependently by PI3K inhibitor but not by MEK inhibitor (figure 9C, D).
Finally, we investigated if MFAP4 influences pro-asthmatic secretory properties of BSMCs. As expected, mRNA expression of CCL11 was strongly enhanced in asthmatic BSMCs compared with healthy cells. Furthermore, CCL11 expression was upregulated in asthmatic BSMCs seeded on MFAP4 compared with cells seeded on control coating (figure 9E). MFAP4-dependent increase in CCL11 expression was inhibited by PI3K but not MEK inhibitor (figure 9F, G).
Discussion
In the present study, we evaluated the role of MFAP4 in OVA-mediated and HDM-mediated allergic asthma models. We showed that OVA and HDM treatments result in elevated serum MFAP4. We further demonstrated that antigen-challenged MFAP4-deficient mice exhibit reduced eosinophilia, goblet cell metaplasia, ASM deposition and AHR compared with WT mice, whereas IgE responses and IL-4 and IL-5 levels remain unchanged. We also showed that MFAP4 is upregulated in asthmatic BSMCs, and that it promotes BSMC integrin-dependent adhesion, proliferation and CCL11 release. Collectively, we suggest MFAP4 as an important contributor to allergic asthma.
MFAP4 levels were increased in serum in OVA-treated mice and in serum and BAL in HDM-treated mice but were decreased in lung homogenates. This suggests that MFAP4 is released to the circulation due to increased matrix turnover and remodelling accompanying disease progression. Elevated MFAP4 expression in asthmatic BSMCs indicates that increased local MFAP4 production may contribute to elevated levels of circulating MFAP4. We showed that MFAP4 is upregulated in asthmatic BSMCs, but other mesenchymal cell types such as vascular SMCs or fibroblasts may constitute another potential source of serum MFAP4. However, this possibility was not explored further.
Correlations between serum MFAP4 and eosinophil numbers and eosinophil chemoattractants support a functional link between MFAP4 and allergic airway disease. Studies that aim to translate this observation by testing serum MFAP4 variation in patients with asthma are thus warranted, in order to assess if MFAP4 may serve as a predictor of eosinophilic asthma in humans.
Eotaxins are potent chemokines playing a crucial role in eosinophil accumulation in asthmatic airways.16 Disturbing eotaxins or their receptor CCR3 impaired lung eosinophil recruitment after antigen challenge, which was accompanied in most, but not all, studies by a decrease in goblet cell metaplasia and AHR.17 ,18 We found reduced levels of eotaxins in the lungs of OVA-treated MFAP4-deficient mice. BSMCs have been previously shown to upregulate CCL11 and other eosinophil-related chemoattractants after stimulation with ECM.19 Our results indicate that MFAP4 contributes to antigen-induced eosinophilia primarily through BSMC-derived CCL11, and can thus indirectly affect goblet cell metaplasia and AHR. However, eotaxins can also be abundantly produced by alveolar macrophages and epithelial cells.20 It is then possible that MFAP4 deficiency results in lowered antigen-induced production of these chemokines from other cell types.
Epithelial transdifferentiation towards mucus-producing cells is strongly influenced by IL-13.21 We found that MFAP4 deficiency resulted in attenuation of goblet cell hyperplasia together with a decrease in local IL-13. However, BSMCs in our in vitro setting did not produce significant amounts of IL-13 (data not shown). We did not investigate MFAP4 effects on other cell types, but both epithelial cells and alveolar macrophages may be relevant sources of IL-13.22 ,23 Considering their expression of RGD-dependent integrins, it is conceivable that MFAP4 may directly regulate IL-13 secretion in these cells. Further studies are needed to clarify these issues.
ASM proliferation is one of the most important causes of airway narrowing and AHR.24 We found that BSMC proliferation was directly influenced by MFAP4, and that lack of MFAP4 partially normalised peribronchial ASM thickness and consequently AHR. Moreover, MFAP4 may also contribute to increased BSMC deposition through eosinophil-dependent hyperplasia.25
Although OVA-induced AHR was strongly attenuated in KO mice, we detected only a tendency towards decreased airway resistance in HDM-treated KO mice. It is possible that severe inflammation after 7-week HDM exposure masked the effects of MFAP4 deficiency on AHR. The two models are mechanistically different; OVA sensitisation together with alum adjuvant with subsequent challenge elicits strong Th2-based adaptive response that is dependent on CD4+ T cells, while repeated local exposure to protease-containing HDM induces more innate response.26 Moreover, even for the same allergen, pathophysiological mechanisms might depend on the duration of the model, as shown for STAT6 involvement in AHR development evident in the acute but not chronic model of OVA exposure (reviewed in27). This underlines that mechanisms driving AHR might be context specific.
We assessed the contributions of central airways and lung parenchyma to the overall AHR increase. The main effect was mediated through changes in tissue damping, a parameter reflecting parenchymal resistance. As the increase in tissue damping is a manifestation of small airway constriction,28 the data suggest that MFAP4-related effects on AHR are caused by inflammation and remodelling in the parenchyma.
There is limited knowledge on expression of integrin receptors on BSMCs. In several studies, α5, αv and β1 subunits were found to be universally expressed, together with moderate expression of α1–α8 and low levels of β3.29 They serve as recognition partners for ECM components such as fibronectin, an established inducer of BSMC proliferation.2 In agreement with existing literature, our data show that BSMCs express high levels of integrin β1 and low levels of integrin β3. We also found high expression of integrin αvβ5 heterodimer, and showed that it serves as a recognition partner for MFAP4. αvβ5 was previously found to activate TGF-β and promote airway remodelling.30 Furthermore, a recent genome-wide study identified ITGB5, a gene coding for integrin β5, to be strongly associated to AHR severity in patients with asthma.31 Our results showed that MFAP4-mediated proliferation was αvβ5-dependent, and thus suggest that interactions between αvβ5 and its ligands, such as MFAP4, contribute to excessive BSMC proliferation seen in asthma.
We detected changes in FAK activation shortly after plating cells onto MFAP4, suggesting that FAK is directly activated by MFAP4-binding integrins. To our knowledge, only one study so far addressed the role of FAK in integrin signalling in ASM, showing that FAK promotes collagen I induced bovine ASM proliferation together with activation of PI3K and ERK.32 Both these pathways regulate growth factor-induced ASM growth, though PI3K seems to be more important in asthmatic ASM.33 Moreover, PI3 signalling has been previously implicated in the regulation of CCL11 expression in ASM, and pharmacological PI3K inhibition has been shown to protect from OVA-induced asthma.34 We demonstrated that MFAP4 effects on proliferation and CCL11 release were inhibited by PI3K but not by ERK pathway blockage, thus identifying a possible mechanism underlying MFAP4-mediated effects.
Integrin signalling and downstream PI3K activation has been shown to promote AP-1 and NF-κB-dependent gene expression (reviewed in Guo and Giancotti35). Binding sites for both these transcription factors lie within the CCL11 promoter region,36 which makes them plausible candidates for mediating MFAP4-related CCL11 upregulation.
Isolated human BSMCs are known to retain their phenotype in vitro, and the phenotypic difference between healthy and asthmatic BSMCs is conserved during isolation and culture. One possible reason might be abnormal calcium homeostasis in asthmatic BSMCs, which is unchanged even after several passages.37
Our study contains several limitations. Human asthma is a complex, heterogeneous disease, manifesting itself in a variety of endotypes.38 The animal models used suggest that MFAP4 contributes to canonical, eosinophilic asthma, but on the basis of the current study we cannot address the possible role of MFAP4 in other subphenotypes, such as neutrophil-rich asthma. Moreover, as our in vitro assays were performed on cells obtained from one donor pair only, they do not provide a full understanding of MFAP4 impact on BSMC behaviour. Further studies, including detailed analysis of patient-derived cells, are thus of interest.
In conclusion, our data indicate that MFAP4 contributes to allergic asthma by promoting airway eosinophilia, AHR and lung remodelling through regulation of ASM proliferation and eotaxin secretion. Our characterisation of this novel pathway involved in asthma pathogenesis might open new research avenues focused on future development of new therapeutic strategies.
Acknowledgments
We thank Vicki Nielsen, Charlotte Skouboe Landré and Ida Tornoe (Institute of Molecular Medicine, Odense, Denmark) for excellent technical assistance. We also thank Christopher Stevenson (Novartis, Horsham, UK) for providing HDM extract and valuable advice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors BP performed experiments, analysed data and wrote the manuscript; AS, HWJ, TT, JBM, NM, JAAP, MHDA, PB participated in data collection and analysis; JV, UH participated in study conception; JBM, JV, UH, GLS, reviewed the manuscript; GLS conceived and supervised the study. BP and GLS are the guarantors responsible for the overall content of the manuscript.
Funding This work was supported by the Lundbeck Foundation, Region of Southern Denmark, Fonden til Laegevidenskabens Fremme, Direktor Jacob Madsen og hustru Olga Madsens Fond, Aase og Ejnar Danielsens Fond, Linexfonden, the Danish Strategic Research Council (the Centre of COPD Research, http://www.cekol.dk) (09-066945) and German Federal Ministry of Education and Research (Infrafrontier grant 01KX1012).
Competing interests AS, UH and GLS have issued a patent: MFAP4 binding antibodies blocking the interaction between MFAP4 and integrin receptors: P1183DK00. JV has not received any financial support in relation to the current manuscript. Outside this work he has received honoraria for advising and presenting from AstraZeneca, Boehringer-Ingelheim, Chiesi, GSK, and Novartis.
Ethics approval Local ethics committee at the Bordeaux Teaching Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.