Article Text

Abstract
Introduction In a subset of patients with asthma, standard-of-care treatment does not achieve disease control, highlighting the need for novel therapeutic approaches. Lebrikizumab is a humanised, monoclonal antibody that binds to and blocks interleukin-13 activity.
Methods LUTE and VERSE were replicate, randomised, double-blind, placebo-controlled studies, evaluating multiple doses of lebrikizumab in patients with uncontrolled asthma despite the use of medium-to-high-dose inhaled corticosteroid and a second controller. Patients received lebrikizumab 37.5, 125, 250 mg or placebo subcutaneously every four weeks. The primary endpoint was the rate of asthma exacerbations during the placebo-controlled period. Analyses were performed on prespecified subgroups based on baseline serum periostin levels. Following the discovery of a host-cell impurity in the study drug material, protocols were amended to convert from phase III to phase IIb. Subsequently, dosing of study medication was discontinued early as a precautionary measure. The data collected for analysis were from a placebo-controlled period of variable duration and pooled across both studies.
Results The median duration of treatment was approximately 24 weeks. Treatment with lebrikizumab reduced the rate of asthma exacerbations, which was more pronounced in the periostin-high patients (all doses: 60% reduction) than in the periostin-low patients (all doses: 5% reduction); no dose–response was evident. Lung function also improved following lebrikizumab treatment, with greatest increase in FEV1 in periostin-high patients (all doses: 9.1% placebo-adjusted improvement) compared with periostin-low patients (all doses: 2.6% placebo-adjusted improvement). Lebrikizumab was well tolerated and no clinically important safety signals were observed.
Conclusions These data are consistent with, and extend, previously published results demonstrating the efficacy of lebrikizumab in improving rate of asthma exacerbations and lung function in patients with moderate-to-severe asthma who remain uncontrolled despite current standard-of-care treatment.
Trial registration numbers The LUTE study was registered under NCT01545440 and the VERSE study under NCT01545453 at http://www.clinicaltrials.gov
- Asthma
- Cytokine Biology
- Asthma Mechanisms
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
Does anti-interleukin-13 (IL-13) treatment improve outcomes in patients with moderate-to-severe asthma?
What is the bottom line?
Lebrikizumab, a potent anti-IL-13 monoclonal antibody, reduces exacerbations and improves lung function in moderate-to-severe asthma patients in a pooled analysis of two phase IIb studies.
Why read on?
To gain understanding of how targeted anti-IL-13 therapy can help asthma patients who remain uncontrolled despite current standard-of-care treatment and how biomarkers can help ensure that patients receive the most appropriate therapy.
Introduction
Asthma is a complex, heterogeneous disease characterised by a variable clinical course, severity and response to treatment.1 ,2 In a subset of patients with asthma, standard-of-care treatment does not adequately control symptoms,1–4 and this is associated with increased healthcare use and high burden of disease.5
Several phenotypes of asthma have been defined2 ,6 ,7 that can be grouped into at least two distinct molecular phenotypes based on the level of expression and activity of type 2 cytokines.8 ,9 Type 2 cytokines, which include interleukin (IL)-13, IL-4 and IL-5, contribute to many aspects of asthma pathology including airway inflammation and hyperresponsiveness.9–11 Of these cytokines, IL-13 has been identified as a central effector cytokine in asthma and recognised as a potential therapeutic target.
Identifying patients whose asthma is type 2-driven is important. Periostin is a matricellular protein that is associated with type 2 inflammation and subepithelial fibrosis in the lung. Bronchial epithelial cells stimulated with IL-13 secrete periostin basolaterally into the extracellular compartment and subsequently periostin accumulates in peripheral blood. Periostin in the circulation therefore serves as a surrogate marker for IL-13 activity in the lung.9 ,11–13
Lebrikizumab is a humanised monoclonal antibody that binds to soluble IL-13 with high affinity and blocks signalling through the active IL-4Rα/IL-13Rα1 heterodimer.8 ,14 ,15 In a previous phase II study (MILLY) in patients whose asthma was inadequately controlled despite treatment with inhaled corticosteroids (ICS), lebrikizumab treatment significantly improved the primary endpoint, FEV1, particularly in patients with higher serum periostin levels.8 The current studies, LUTE and VERSE, were designed to evaluate the efficacy and safety of different doses of lebrikizumab in a larger number of patients with moderate-to-severe asthma, treated for a longer duration and evaluating the rate of exacerbations as the primary endpoint.
Methods
Study design
LUTE and VERSE were replicate, randomised, multicentre, double-blind, placebo-controlled studies (NCT01545440 and NCT01545453). The studies were initially designed to enrol approximately 1400 patients each and to include a 52-week double-blind treatment period (see below for study modification). Patients were randomised in a 1:1:1:1 ratio to receive lebrikizumab 37.5, 125, 250 mg, or placebo subcutaneously every four weeks. Randomisation was stratified by baseline serum periostin level, history of asthma exacerbations within the last 12 months and baseline asthma medications. All patients remained on their standard-of-care therapy that consisted of 500–2000 μg/day ICS therapy (fluticasone propionate dry powder inhaler (DPI) or equivalent) and a second eligible asthma controller medication.
Study population
Patients aged 18–75 years with uncontrolled asthma despite daily use of 500–2000 μg/day of fluticasone propionate DPI or equivalent and a second asthma controller medication were included in the studies.1 ,16 Second controller medications included long-acting β2-agonists, leukotriene receptor antagonists, long-acting muscarinic antagonists or theophylline.
Detailed inclusion and exclusion criteria are included in the online supplementary material. Briefly, inclusion criteria included diagnosis of asthma ≥12 months, acute bronchodilator response (≥12% relative improvement) and pre-bronchodilator FEV1 40–80% of predicted. Uncontrolled asthma was defined as an Asthma Control Questionnaire-5 score ≥1.5 and at least one of the following: symptoms >2 days/week, night-time awakenings ≥1 time/week, use of a short-acting β2-agonist as rescue medication >2 days/week or interference with normal daily activities. During the screening period, patients were asked to report adherence with background controller medications, and only patients who reported adherence with controller medications could be considered for randomisation. Patients continued their stable background asthma controller medications for the duration of the studies. There was no run-in period as patients remained on their own stable medications and randomised treatment with lebrikizumab or placebo started on day 1. Patients were excluded if they had received maintenance oral corticosteroid treatment within the previous three months or treatment with systemic corticosteroids within the previous four weeks for any reason.
Efficacy and safety assessments
The primary endpoint was the rate of asthma exacerbations during the placebo-controlled period. Due to early termination of dosing, the observation period was variable for each individual patient; see study modification below. An asthma exacerbation was defined as new or increased asthma symptoms that led to treatment with systemic corticosteroids or to hospitalisation. Treatment with systemic corticosteroids was defined as oral, intravenous or intramuscular corticosteroid treatment for ≥3 days or an emergency room visit with ≥1 dose of intravenous or intramuscular corticosteroids. Asthma exacerbations were assessed at each study visit by the investigator using directed questions to assess whether the patient had experienced any asthma exacerbations since the last visit. It was prespecified that the primary and all secondary endpoints would be evaluated separately in the periostin-high and periostin-low groups (based on a cut-point of 50 ng/mL). Spirometry (pre-bronchodilator and post-bronchodilator) was assessed throughout the study, and methods are described in the online supplementary material. A peak flow/eDiary device was used for once-daily measurement of peak expiratory flow (between 5:00 and 11:00) and recording of asthma rescue medication and controller use.
Prespecified secondary endpoints were relative change in pre-bronchodilator FEV1 from baseline, time to first asthma exacerbation during the placebo-controlled period, change from baseline in the asthma-specific health-related quality-of-life measure, Asthma Quality-of-Life Questionnaire (standardised; (AQLQ(S))), change in asthma rescue medication use from baseline, rate of urgent asthma-related healthcare use (ie, hospitalisations, emergency department visits and acute care visits) during the placebo-controlled period.
Safety endpoints were the rate and severity of adverse events (AEs) during the placebo-controlled and follow-up periods and the incidence of antitherapeutic antibodies (ATAs) during the study relative to baseline.
Study modification
A host cell protein impurity (PLBL2)17 was identified after the initiation of the studies. This required manufacturing process changes to the drug product. As a consequence, the studies were no longer considered pivotal studies and the protocols were amended from phase III to phase IIb. Upon conversion to phase IIb, it was decided that each study would enrol 225 patients from approximately 70 sites located in the USA (for a total of 450 patients). The planned placebo-controlled period was of 28–52 weeks' duration, with a variable number of doses administered depending on when the patients enrolled in the studies. After the treatment period, patients were followed for safety for 24 weeks (>5 half-lives of the drug) following the last dose of study drug, including those who discontinued treatment early (see online supplementary figure S1). The protocols further specified that data from the two studies would be pooled for analysis and an internal administrative review of the unblinded data would inform the design of subsequent phase III clinical studies.
Coincident with the administrative data readout, results from an immunoassay designed to measure immune response to the host cell protein impurity demonstrated that nearly all lebrikizumab-treated patients showed an immune response. At that point, a decision was made to discontinue dosing early as a precautionary measure to avoid patients’ further exposure to the impurity-containing study drug material. All patients were asked to complete the safety follow-up period.
Statistical analyses
Data obtained from LUTE and VERSE were pooled for analysis. Based on the sample size and treatment duration as originally planned in the phase IIb protocols, the combined sample size of approximately 450 patients (56 patients per treatment arm per periostin subgroup) was estimated to provide 70% power to detect a 50% reduction in the rate of exacerbations within each periostin subgroup for a given lebrikizumab dose group compared with placebo. Calculations were based on a Poisson regression model and two-sided test at the α=0.20 level with the assumption of an average rate of 0.6 exacerbations per patient-year in the placebo arm, a 20% dropout rate and 20% Poisson over-dispersion.
As prespecified in the protocols, analysis of the primary efficacy and all secondary efficacy endpoints were performed separately in periostin-high (≥50 ng/mL) and periostin-low (<50 ng/mL) subgroups.
Because dosing was stopped early, all efficacy analyses were considered at that point to be exploratory, and results for efficacy outcomes were primarily summarised in a descriptive fashion. Point estimates of treatment effect were provided along with 95% CIs. P values for specific comparisons of interest were included, as supportive information, without adjustments for multiplicity comparisons. Owing to incomplete data at later time points, the primary time point used for comparison of the lebrikizumab arms versus the placebo arm for secondary or exploratory outcomes was week 12.
Results
Patients
The pooled population included a total of 463 patients. The LUTE study randomised 258 (56%) patients and the VERSE study 205 (44%) patients. At the time of dosing termination, patients discontinued the placebo-controlled treatment period and entered into the 24-week safety follow-up period.
The baseline demographic and disease characteristics were similar across treatment groups (table 1).
Patient baseline characteristics
Because dosing was terminated early, not all patients had the opportunity to participate in the placebo-controlled treatment period for the minimum duration (ie, seven doses of study drug over 28 weeks) as specified in the amended protocols.
The median duration of treatment was 24.1 weeks, and the median time spent in the study, including the safety follow-up period, was 44.1 weeks (table 2). Patients received a median of six (range 1–12) doses of study drug during the placebo-controlled period (table 2).
Time in study and drug exposure
Efficacy
Asthma exacerbations
For the primary outcome, lower exacerbation rates were observed in the lebrikizumab treatment groups compared with placebo (table 3). The exacerbation rate reduction compared with placebo was more pronounced in periostin-high patients than in periostin-low patients (table 3). In periostin-high patients, there was a 60% (95% CI 18% to 80%) reduction in the rate of exacerbations for the lebrikizumab dose groups combined compared with placebo in periostin-high patients. In the periostin-low patients, only a 5% (95% CI −81% to 47%) reduction was observed (table 3).
Protocol-defined asthma exacerbations during the placebo-controlled period
There was no clear dose–response for lebrikizumab on exacerbations. Compared with placebo, the exacerbation rate was reduced by 81% (95% CI 35% to 97%), 77% (95% CI 26% to 95%) and 22% (95% CI −62% to 63%) in periostin-high patients and by 33% (95% CI −53% to 73%), −17% (95% CI −141% to 42%) and 5% (95% CI −114% to 59%) in periostin-low patients in the lebrikizumab 37.5, 125 and 250 mg groups, respectively (figure 1). Kaplan–Meier plots of time to first exacerbation are shown in the online supplementary material (see online supplementary figure S2) as are exacerbation data for each trial (see online supplementary table S1).

Rate of asthma exacerbations during the placebo-controlled period.
Lung function
The mean relative change in FEV1 for the pooled lebrikizumab group versus placebo from baseline to week 12 in periostin-high patients was 9.1% and 2.6% in periostin-low patients. By dose, in periostin-high patients, this was 6.8% (95% CI −0.7% to 14.4%) in the lebrikizumab 37.5 mg group, 10.7% (95% CI 0.6% to 20.7%) in the lebrikizumab 125 mg group and 10.1% (95% CI 1.3% to 18.9%) in the lebrikizumab 250 mg group (figure 2). In comparison, in periostin-low patients, the mean relative change from baseline in FEV1 versus placebo was −1.9% (95% CI −8.3% to 4.6%) in the lebrikizumab 37.5 mg group, 2.2% (95% CI −3.4% to 7.9%) in the lebrikizumab 125 mg group and 7.2% (95% CI −0.7% to 15.2%) in the lebrikizumab 250 mg group.

Mean (SE) percentage change in FEV1 from baseline up to week 12.
Secondary outcomes
Other secondary outcomes at week 12 are summarised in table 4 and described in more detail in the online supplementary material. Briefly, there was evidence of increased time to first exacerbation with lebrikizumab treatment in periostin-high patients, especially in the 37.5 and 125 mg groups, and a similar effect reducing the rate of urgent asthma-related healthcare use in periostin-high patients. For other outcomes including the patient-reported AQLQ, there was little indication of treatment differences but CI were wide, thus limiting interpretation.
Secondary and exploratory efficacy and pharmacodynamic endpoints
Biomarkers
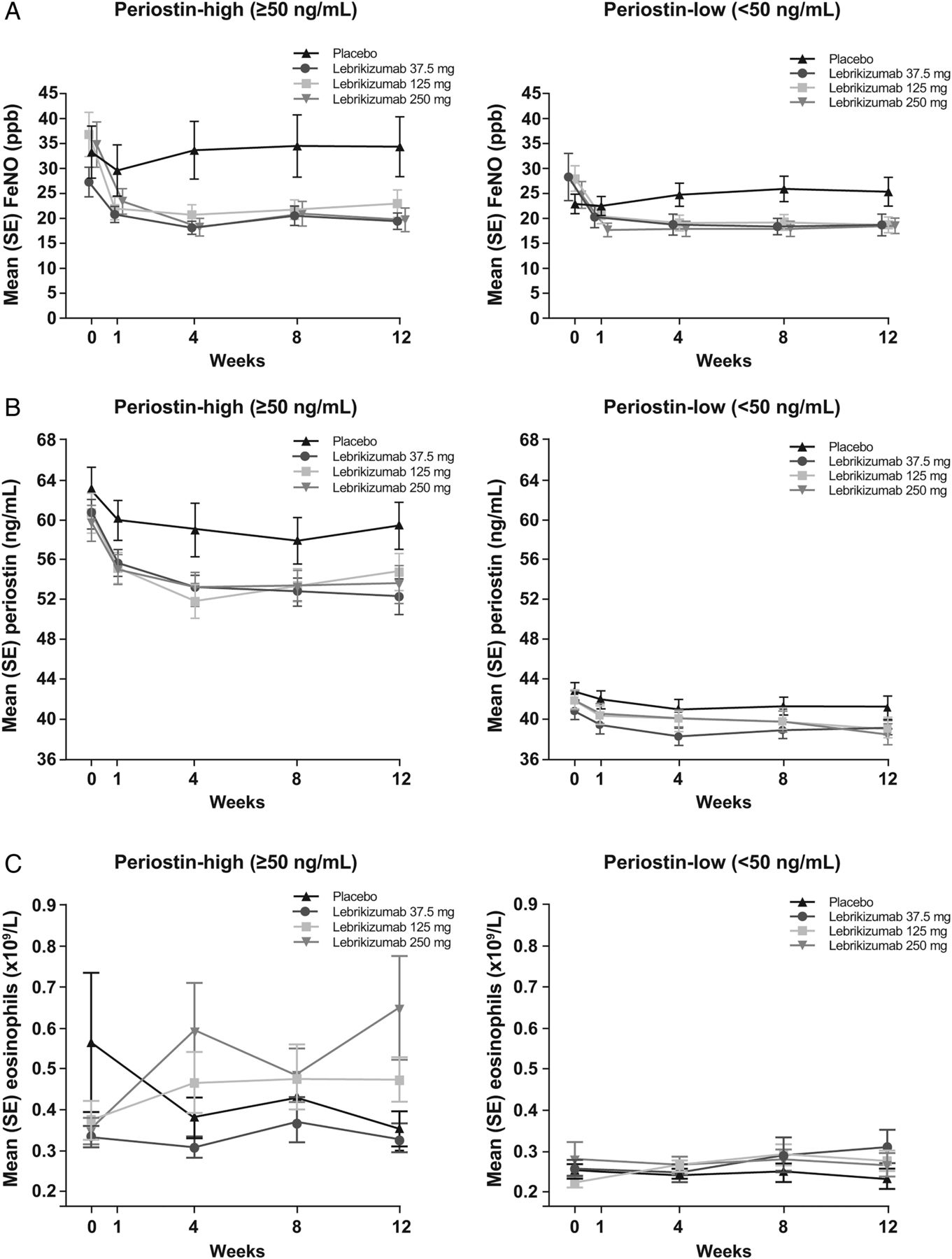
Figure 3 shows the average change in biomarker values (FeNO, blood eosinophils and serum periostin) at each study visit through to week 12 by treatment group in periostin-high and periostin-low patients. More detailed description of methods and results is presented in the online supplementary material.

{kind=link}
{kind=link}
{kind=link}
Mean values for (A) FeNO, (B) periostin and (C) blood eosinophils over 12 weeks.
Preliminary analyses of exacerbation and lung function data stratified by baseline FeNO and blood eosinophil count are provided in the online summary (see online supplementary table S3). Raised FeNO (≥21 ppb) and blood eosinophils (≥240 cells/µL) were both predictive of treatment response to lebrikizumab.
Pharmacokinetics
Pharmacokinetic analyses showed that lebrikizumab concentrations in serum increased roughly proportionally with the dose levels. Overall, the pharmacokinetics of lebrikizumab were as expected based on data from previous trials. The observed mean (SD) of trough concentrations at week 12 were 4.0 (±1.9), 14.2 (±6.4) and 26.5 (±11.1) μg/mL for the 37.5, 125 and 250 mg dose groups, respectively. As expected, this shows overlap in the ranges of exposure for the 125 and 250 mg dose.
Safety
Among all patients, 9.7% of patients discontinued treatment prior to the discontinuation of dosing, with similar rates across treatment groups (see online supplementary figure S1). The incidence of AEs was generally similar across the treatment groups (table 5). In total, 30 serious adverse events (SAEs) were reported in 21 patients during the entire study (including the safety follow-up period) and 12 SAEs were reported in nine patients during the placebo-controlled part of the study (see online supplementary material for details). Injection site reactions were numerically higher in the lebrikizumab 125 and 250 mg dose groups compared with the lebrikizumab 37.5 mg group or placebo (20.5%, 20.3%, 11.1% and 6.0%, respectively). There were 11 AEs leading to withdrawal of study drug, 8 in lebrikizumab-treated patients and 3 in placebo (see online supplementary material for details). AEs leading to discontinuation were varied and did not suggest a safety concern.
Adverse events (AEs) reported during the entire study
Five neoplasms (capturing benign, malignant and unspecified neoplasms including cysts and polyps) were reported during the study, none of which were considered related to study drug (see online supplementary material for details). Two non-serious hypersensitivity reactions were reported in the placebo group and one each in the lebrikizumab 125 and 250 mg groups. There were no cases of anaphylaxis, anaphylactoid or serious hypersensitivity reactions reported during the study. ATAs and antibody response to the impurity identified (PLBL2) were detected but not associated with any AE (more details are available in the online supplementary material).
Discussion
In two replicate studies in patients with moderate-to-severe asthma uncontrolled despite ICS therapy and an additional controller, lebrikizumab administered subcutaneously every four weeks reduced asthma exacerbation rate by 60% (95% CI 18% to 80%) compared with placebo in periostin-high patients and by 5% (95% CI −81% to 47%) in periostin-low patients. In addition, lebrikizumab improved lung function, as measured by change in FEV1, in periostin-high patients. However, the wide CIs in both exacerbations and lung function measures must be acknowledged. Despite these improvements in lung function, lebrikizumab treatment did not lead to clinically meaningful placebo-corrected improvements in asthma symptoms or quality of life, potentially due to the limited power of the studies for these endpoints. Furthermore, lung function does not always correlate with subjective measures in clinical trials and previous trials of biological therapy that have shown improvements in symptoms or quality of life have reduced background therapy or have involved larger patient populations.18 ,19 Lebrikizumab was generally well tolerated and no clinically important safety signals were observed. It is also notable that, besides periostin, both blood eosinophil count and FeNO were predictive of clinically meaningful treatment benefit using biomarker high and low groups defined by the median values for these biomarkers.
In the previous phase II (MILLY) study in moderate-to-severe asthma, lebrikizumab significantly improved lung function, with the greatest changes in periostin-high patients. There was a non-significant trend for a lower exacerbation rate through 24 weeks with lebrikizumab treatment compared with placebo.8 The placebo-corrected rate of exacerbations was reduced by 67% in periostin-high patients and by 29% in periostin-low patients.8 The results from the combined phase IIb studies, LUTE and VERSE, extend the findings described previously, in a larger population with more severe asthma. The results from these studies also support the finding that baseline periostin level can be predictive of lebrikizumab treatment benefit. Of note was that periostin-low patients in the placebo group experienced fewer exacerbations than periostin-high patients in the placebo group in the combined phase IIb studies, LUTE and VERSE. Larger phase III trials, currently underway, are required to confirm over longer duration of treatment the effects observed in this study on the rate of asthma exacerbations and lung function, as well as to further investigate whether these improvements can translate into symptom benefits for patients.
The IL-13 contribution to the pathophysiology is not limited to patients with a history of exacerbations. Accordingly, in neither the current studies nor the previous phase II study (MILLY) was a history of exacerbation required. In the combined phase IIb studies, LUTE and VERSE, approximately 52% of patients had not experienced an exacerbation in the previous year. We observed no dose–response for lebrikizumab on exacerbation rates; paradoxically, the lowest rate reduction was observed in the highest dose group. The higher rate of exacerbations in the pooled 250 mg lebrikizumab group is largely driven by a higher number of exacerbations in the periostin-high arm of the VERSE trial (see online supplementary table S1). The reason for this higher rate is unclear. The small sample sizes and short median observation period also contributed to the decrease in precision in the analyses.
The apparently smaller rate reduction in the 250 mg group is unexpected given that a previous study in a similar patient population8 showed efficacy at 250 mg comparable to that of 125 mg in this study. Furthermore, data from secondary efficacy endpoints and biomarkers reported here do not show a lower response at 250 mg compared with 125 mg. While there was approximately a twofold difference in the average exposure of the 125 and 250 mg dose groups, there is a partial overlap in the concentration ranges. As there was no apparent relationship between lebrikizumab concentration and exacerbation rate, it suggests a potential plateau in the dose–response. Improvement in FEV1 was less pronounced in the lebrikizumab 37.5 mg dose cohort compared with the 125 and 250 mg dose cohorts in the periostin-high group, suggesting the 37.5 mg dose was a partially effective dose. The FEV1 response to lebrikizumab was rapid with improvement within 1 week and near maximal improvements reached by week 4—a pattern also seen in the previous phase II study (MILLY).8 The mechanism for this rapid response is unclear, although it has been suggested previously that enrolment in a clinical trial may be associated with rapid improvements simply due to improved adherence to background therapy. However, in this study, the patients completed a screening period lasting at least 14 days, during which patients were assessed for adherence with their background asthma therapy and the degree of asthma control provided by their standard-of-care asthma medications. Patients whose symptoms remained uncontrolled during this screening period despite adherence with controller medicines were eligible to participate. Therefore, placebo-corrected improvements in lung function seen during week 1 are unlikely to be due to improved adherence to therapy.
Changes in peripheral blood eosinophil counts have been reported previously8 ,20 and may reflect blocking of IL-13 activity. The increased eosinophil counts in blood may be due to decreased migration from blood to the airways due to reduced chemotaxis.21 ,22
Our study has several limitations. The change in study design after initiation of the studies resulted in variation of study drug exposure among patients; however, on average, the exposure was balanced between the treatment arms. Some patients were treated for a relatively short period of time (eg, 1–2 doses) and drug exposures did not reach steady state in all patients. The sample size of 463 patients may not be sufficient to completely understand the dose–response—especially considering exacerbations are relatively infrequent events. In spite of these limitations, these studies confirm the role of periostin as a predictor of response to IL-13-targeted therapy in moderate-to-severe, uncontrolled asthmatics. Neutralising IL-13 activity in periostin-high patients reduced the rate of acute exacerbations and improved FEV1.
In addition, these studies further confirm the safety profile of lebrikizumab. Injection site reactions were more common in patients with the higher doses of lebrikizumab than with the lower dose or placebo. The assay for antibodies against the host cell impurity PLBL2 detected a response in the majority of lebrikizumab-treated patients; however, the clinical significance of anti-PLBL2 antibodies is not known. No clinically important safety signals were identified in this study.
The results of these studies, while providing further evidence of the safety and efficacy of lebrikizumab in patients with moderate-to-severe uncontrolled asthma, need to be confirmed in larger randomised, double-blind, placebo-controlled phase III clinical studies, which are currently underway.
Acknowledgments
LUTE and VERSE were funded by Genentech, a member of the Roche Group. Support for third-party writing assistance for this article, furnished by Jonathan Brennan, PhD, of MediTech Media, was provided by F. Hoffmann-La Roche Ltd.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors All authors made substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work. All authors were involved with drafting the work or revising it critically for important intellectual content and all have approved the version to be published.
Competing interests YZ, SKF, MC, WSP, EM, HS, CTJH, RM, SG, RD, DM, JO, JGM and KY are all employees of Genentech, a member of the Roche Group. NAH has received honoraria for serving on advisory board or consultant for Genentech, Roche, Sunovion, Pfizer and Novartis. His institution has received research grants on his behalf from Genentech, GSK, BI, Mylan, Sunovion, Pearl, Pfizer. MN has received honoraria as a speaker for Astrazeneca. PK's institution received a research grant provided by Genentech to Clinical Research Center LLC on his behalf. JC has received honoraria as a speaker for Genentech and has received research grants from Genentech.
Patient consent Obtained.
Ethics approval Approved by applicable institutional review board or independent ethics committee at each participating centre.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves