Article Text

Statistics from Altmetric.com
JPC: A 63-year-old Caucasian man was seen in clinic having been referred by his general practitioner with a 10 -week history of progressive exertional dyspnoea, associated with dry cough and retrosternal discomfort over the preceding fortnight. There was no history of fever, weight loss, anorexia, haemoptysis, dysphagia or other localising systemic symptoms. His past medical history included psoriatic arthropathy, atrial fibrillation, hypertension and gastro-oesophageal reflux; of note, there was no recent history of palpitations or worsening peripheral oedema, while his joint disease remained quiescent. Current medications (all long-standing) included methotrexate, omeprazole, bisoprolol, aspirin, ramipril and simvastatin. He was a lifelong non-smoker and retired solicitor with no occupational risk factors for lung disease.
On examination he was afebrile with no palpable lymphadenopathy or clubbing. Cardiovascular examination demonstrated rate-controlled atrial fibrillation only; abdominal examination was normal. Respiratory examination revealed dull percussion note and reduced breath sounds at the right lung base, with normal resting oxygen saturations. Chest radiograph and thoracic ultrasound confirmed the presence of a large right-sided pleural effusion. Routine bloods including full blood count, clotting studies, renal function, liver function and serum calcium were unremarkable. I performed a diagnostic and therapeutic pleural aspiration, draining 1500 mL of straw-coloured fluid with no complications. Laboratory analysis showed the effusion to be an exudate (protein 46 g/L, lactate dehydrogenase (LDH) 961 IU/L, glucose 0.4 mmol/L); pleural fluid culture and cytology (including flow cytometry) were negative.
NMR: The presence of a cytology-negative exudative pleural effusion of unknown aetiology in a previously well individual requires urgent further investigation. The low pleural fluid glucose and high LDH may imply infection, although these findings are non-specific and can also be seen in the context of chronic inflammation or malignancy; furthermore, the clinical history and negative culture go against an infective cause. The presence of an inflammatory arthropathy may prompt consideration of whether an underlying connective tissue or autoimmune disease is responsible; methotrexate is also associated with pleural effusion formation. In the first instance, a contrast-enhanced CT scan of the thorax (staging protocol, ideally taken in the venous phase to allow assessment of the pleura) should be performed. Unless this demonstrates an indisputable cause for the effusion or a more suitable diagnostic route, the patient will need to have pleural biopsies taken with the aim of making a definite diagnosis—it is worth remembering that neither negative pleural fluid cytology nor an apparently normal CT scan can definitively exclude an underlying pleural malignancy.
JPC: The patient's CT scan revealed a moderate right-sided pleural effusion that enhanced peripherally but also appeared to contain a heterogeneously enhancing curvilinear opacity, potentially of vascular origin. Subsequent MRI thorax excluded an occult ongoing intrapleural bleed. Smooth visceral and parietal pleural thickening was noted, but there were no features that would be consistent with a malignant process such as nodularity, chest wall invasion or mediastinal involvement. The lung parenchyma, mediastinum and upper abdominal organs appeared otherwise unremarkable.
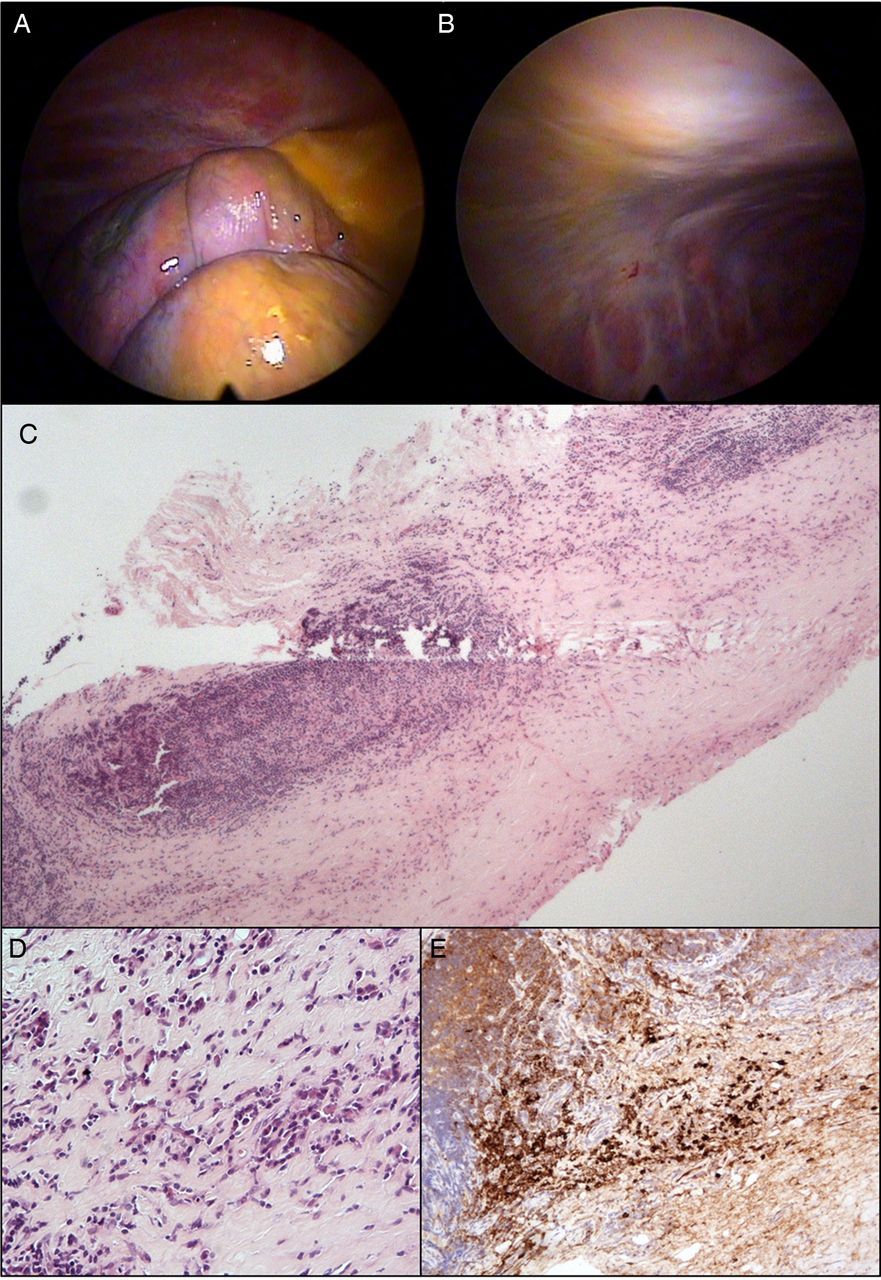
Local anaesthetic thoracoscopy was performed in order to obtain pleural tissue for histological analysis. A further 2200 mL of straw-coloured fluid was drained from the right hemithorax; visual inspection of the pleural cavity identified two mobile mass lesions with an apparently fatty consistency adjacent to the diaphragm, alongside diffuse pleural thickening involving both parietal and visceral (right lower lobe) surfaces (figure 1A,B). Full-thickness pleural biopsies were taken from the extensive parietal thickening at multiple sites using a two-port technique; biopsies were also obtained separately from the two mass lesions. Talc poudrage was not performed, meaning the patient could be discharged home the same day after chest radiograph and thoracic ultrasound confirmed re-expansion of the right lung.

{kind=link}
Images (A) and (B) taken during local anaesthetic thoracoscopy demonstrate inflammatory pleuritis and diffuse thickening involving the parietal and visceral surfaces; subsequent pleural biopsies demonstrate histological changes consistent with immunoglobulin G4 (IgG4)-related pleural disease including: (C) and (D) fibrous thickening and prominent lymphoplasmacytic infiltrate (H&E); and (E) strongly positive staining for IgG4-positive plasma cells.
SJD: Biopsies from the intrathoracic mass lesions demonstrated fibrin only with some entrapped degenerate cells of inflammatory origin and peripheral macrophages; no malignant cells were seen. The pleural biopsies (figure 1C–E) showed features consistent with chronic inflammation and fibrinous pleuritis. In some areas, a single layer of histiocytes was visible at the surface, but no malignant cells were identified. The inflammatory change was rich in lymphocytes with lymphoid follicle formation focally and, in addition, a highly prominent population of plasma cells that stained positive for immunoglobulin G4 (IgG4). There were no features of haematological malignancy identified, with kappa and lambda light chain immunohistochemistry excluding a clonal plasma cell population. No obliterative phlebitis was observed. These findings raised the possibility of the pleural inflammation and thickening being secondary to IgG4-related disease and prompted referral of the histology for a second expert opinion.
ACB: A review of the pleural tissue received demonstrated fibrous thickening with, in areas, a prominent lymphoplasmacytic chronic inflammatory infiltrate. Active myofibroblasts were present but storiform pattern fibrosis was not identified. Immunohistochemistry revealed a mean of 73 IgG4-positive plasma cells per high-power field, with a ratio of IgG4-positive to IgG-positive plasma cells >40%. While the features were not diagnostic of IgG4-related disease based on histopathological evidence alone, they would be in keeping with ‘probable histological features of IgG4-related disease’ according to the Boston criteria.1
JPC: The patient was reviewed in clinic to discuss the pleural biopsy results and plan further investigations. Once again he reported no recent or previous history of abdominal symptoms. Serum and urine electrophoresis was normal, while serum IgG4 level was elevated at 2.84 g/L (normal range 0.1–1.3 g/L). Autoimmune profile including antinuclear antibody, complement and rheumatoid factor was unremarkable; serum amylase was also normal. Chest radiograph and thoracic ultrasound confirmed further recurrence of the patient's right pleural effusion, although not to the point whereby repeat aspiration for symptomatic benefit was required. Completion imaging with CT scan of the abdomen and pelvis did not demonstrate any further pathological abnormality. The patient's case was subsequently discussed at the regional IgG4-related disease multidisciplinary team meeting where in the context of clinical, biochemical, radiological and histological correlation a diagnosis of IgG4-related pleural disease was agreed.
NMR: Having initially been recognised in association with autoimmune pancreatitis nearly two decades ago, IgG4-related disease is now increasingly acknowledged as being a multisystem fibroinflammatory disorder that can affect almost any organ system including the lungs.2 The most common intrathoracic manifestation of IgG4-related disease is hilar or mediastinal lymphadenopathy, seen in up to 80% of patients. Pulmonary involvement can be extremely variable with features including solid nodules or masses, bronchiectasis, bronchovascular involvement and interstitial lung disease all described either in isolation or combination. Pleural involvement in the form of effusions and/or thickening (with or without underlying lung parenchymal involvement) is also increasingly recognised in the literature base.3 The key differential diagnosis in this particular case was a lymphoproliferative disorder; the absence of clonality on either pleural fluid flow cytometry or pleural biopsy histology was reassuring in this regard, as was the absence of lymphadenopathy on full-body CT imaging.
ACB: Diagnosing IgG4-related disease is frequently challenging, with elevated tissue IgG4-positive plasma cell numbers helpful but not specific. Instead, it is necessary to identify characteristic histopathological features1 that are usually consistent regardless of the site of disease. The three key histopathological features are dense lymphoplasmacytic infiltration, storiform pattern fibrosis and obliterative phlebitis (venulitis), although it is common for not all of these to be present within a tissue sample. However, the level of confidence for a diagnosis of IgG4-related disease increases if at least two—and ideally all three—morphological features are present. The diagnosis can then be further supported by the demonstration of prominent IgG4-positive plasma cells and a ratio of IgG4-positive to IgG-positive plasma cells of >40%. Other features may include the variable presence of eosinophils within the tissue. As with any diagnosis, the clinical history, serology, radiology and histology should be carefully correlated to ensure consistency across these characteristics.
EB: IgG4 is ordinarily the least abundant subclass of IgG in healthy individuals and has traditionally been regarded as having an anti-inflammatory role, being unique among the subclasses of IgG in not activating complement or forming immune complexes. Nonetheless, it is recognised as being potentially involved in a number of autoimmune conditions including forms of pemphigus and membranous glomerulonephritis; and separate to this it underlies the collection of related disorders that have now bracketed under the internationally recognised term of ‘IgG4-related disease’.4
The pathogenesis of IgG4-related disease is at this point in time poorly understood, perhaps unsurprising since we have limited understanding of the role IgG4 even plays in normal health. Furthermore, it is unclear whether the excessive levels of IgG4 seen in affected patients are pathogenic and destructive in themselves, or simply an exaggerated response to another unidentified primary immunological stimulus. Proposed triggers for IgG4-related disease include genetic and human leucocyte antigen-associated susceptibility, autoimmunity, an abnormal allergic response and molecular mimicry by an infective agent. The subsequent development of the condition is thought to be driven by an overabundance of Th2 and regulatory T cells in the affected tissues, a key distinction from the vast majority of autoimmune conditions where the function of regulatory T cells is classically either impaired or downregulated.
Being an only recently described entity, we have limited information about the epidemiology of IgG4-related disease. In addition, a lack of awareness of the condition alongside the ability of IgG4-related disease to mimic a number of better-known diagnoses (eg, primary sclerosing cholangitis in the biliary system or sarcoidosis in the lung) means it may have been and still remain under-recognised. The data available indicate that IgG4-related disease is most frequently seen in male patients over the age of 50 years2—a striking contrast to most autoimmune disease that predominates in a young female population—with autoimmune pancreatitis remaining the best recognised and studied presenting feature. Data (unpublished) from the Oxford IgG4-related disease patient cohort and that of others5 show an increased risk of malignancy both at diagnosis and during follow-up compared with age-matched and sex-matched controls, the reasons for which are not understood at this point in time. As such, the presence of malignancy in patients with suspected IgG4-related disease should be actively excluded at the time of diagnosis and monitored for during subsequent treatment and follow-up.
It is important to recognise that IgG4-related disease should be a diagnosis made through the identification of specific histopathological findings placed in an appropriate clinical context. Relying on serum IgG4 alone may result in underdiagnosis since a number of patients with biopsy-proven IgG4-related disease will have normal serum levels; and indeed overdiagnosis as approximately 5% of healthy controls can have elevated serum IgG4 levels at any point in time. Nonetheless, making the correct diagnosis is crucial since IgG4-related disease is typically steroid-responsive in the absence of established tissue fibrosis with symptomatic resolution and improved function of the affected organ(s). It remains unclear whether early diagnosis and intervention has an impact on long-term outcomes, and this will only become apparent over time and with greater clinical experience.
Steroid dosing varies according to local practice, but initial treatment regimens with oral prednisolone of between 30 mg and 1 mg/kg daily are commonly used. Assuming a favourable response, this can be gradually weaned over subsequent months. In resistant or relapsing cases, despite steroid therapy additional immunomodulatory medications such as rituximab, azathioprine and mycophenolate can be used, although this is not on the basis of any randomised trial data.
JPC: Our patient was informed of the diagnosis of IgG4-related pleuritis and started on oral prednisolone at a dose of 40 mg daily, weaning gradually according to clinical and radiological response. He remains well on a low-maintenance dose of prednisolone after 8 months, with near-total resolution of his previously reported respiratory symptoms. Serum IgG4 levels have returned to within normal limits, while there is evidence of reduction in both pleural thickening and effusion volume on serial imaging with no features to suggest further organ system involvement by his IgG4-related disease.
Footnotes
-
Contributors The article was conceived by JPC, ELC, EB and NMR. JPC and ELC were responsible for draft preparation and revision. All authors were involved in critical appraisal of each draft and approval of the final submitted manuscript. All authors contributed to the patient's clinical care. JPC is the lead author of this article; JPC and NMR are responsible for the overall content as guarantors.
-
Funding IP is the recipient of a European Respiratory Society Fellowship (LTRF 2013-1824). EB and NMR are funded by the NIHR Oxford Biomedical Research Centre.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Informed written consent was obtained from the patient prior to the production of this article.
-
Provenance and peer review Not commissioned; externally peer reviewed.