Article Text

Abstract
Background Activation and differentiation of fibroblasts into contractile protein-expressing myofibroblasts and their acquired apoptosis-resistant phenotype are critical factors towards the development of idiopathic pulmonary fibrosis (IPF), a fatal disease characterised by distorted pulmonary structure and excessive extracellular matrix (ECM) deposition. The molecular mechanisms underlying these processes in IPF remain incompletely understood. We investigated the possible implication of aberrant overexpression and activity of histone deacetylases (HDACs) in IPF.
Methods We analysed lung tissues from patients with sporadic IPF (n=26) and non-diseased control lungs (n=16) for expression of class I and II HDACs. Primary IPF fibroblasts were treated with HDAC inhibitors (HDACi) LBH589 or valproic acid (VPA).
Results Compared to control lungs, protein levels of class I (HDAC1, HDAC2, HDAC3, HDAC8) and class II HDACs (HDAC4, HDAC 5, HDAC 7, HDAC 9) were significantly elevated in IPF lungs. Using immunohistochemistry, strong induction of nearly all HDAC enzymes was observed in myofibroblasts of fibroblast foci and in abnormal bronchiolar basal cells at sites of aberrant re-epithelialisation in IPF lungs, but not in controls. Treatment of primary IPF fibroblasts with the pan-HDACi LBH589 resulted in significantly reduced expression of genes associated with ECM synthesis, proliferation and cell survival, as well as in suppression of HDAC7, and was paralleled by induction of endoplasmic reticulum stress and apoptosis. The profibrotic and apoptosis-resistant phenotype of IPF fibroblasts was also partly attenuated by the class I HDACi VPA.
Conclusions Aberrant overexpression of HDACs in basal cells of IPF lungs may contribute to the bronchiolisation process in this disease. Similarly, generation and apoptosis resistance of IPF fibroblasts are mediated by enhanced activity of HDAC enzymes. Therefore, pan-HDAC inhibition by LBH589 may present a novel therapeutic option for patients with IPF.
- Idiopathic pulmonary fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
Although altered activity and expression of histone deacetylases (HDACs) have been reported in various pathological conditions, such as cancer or COPD, no data are currently available about the regional expression patterns of HDAC enzymes in idiopathic pulmonary fibrosis (IPF), a fatal lung disease for which no cure exists to date.
What is the bottom line?
This study shows for the first time that both myofibroblasts of fibroblast foci and bronchiolar basal cells of IPF lungs reveal ‘cancer-like’ overexpression of class I and class II HDAC enzymes, which may explain their progressive expansion as well as apoptosis resistance in this disease.
Why read on?
This study demonstrates for the first time that the pan-HDAC inhibitor LBH589/panobinostat, which is a proven antitumour agent, is capable of significantly downregulating collagen I biosynthesis and antiapoptotic genes in IPF fibroblast populations, and may thus potentially stop fibrotic remodelling in IPF.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a fatal lung disease characterised by destruction of alveolar architecture, abnormal fibroblast proliferation and excessive collagen deposition. Myofibroblasts are the primary collagen-producing cells in IPF, and their accumulation within pathological lesions called fibroblast foci (FF) is a key feature of IPF lungs.1 Moreover, it has been suggested that IPF fibroblasts and myofibroblasts possess a cancer-like, ‘malignant’ phenotype,2 with enhanced resistance to apoptosis due to increased expression of antiapoptotic and survival-related proteins, such as phosphatidylinositol-3-kinase-γ3 or survivin.4 However, the precise mechanisms of such apoptosis resistance remain unclear.
Structural changes of the chromatin, including alterations in the histone acetylation/deacetylation balance, have been suggested to underlie altered gene expression patterns in various pathological conditions, including cancer,5 cardiac hypertrophy6 and COPD.7 Histone deacetylases (HDACs) are enzymes that remove acetyl moieties from ε-N-acetylated lysine residues in histones, resulting in chromatin condensation and epigenetic repression of gene transcription. HDACs also catalyse deacetylation of many non-histone proteins, such as the tumour suppressor p53, resulting in inhibition of its (pro-apoptotic) transcriptional activity.8 ,9 In line with their profound antiapoptotic activity, HDACs are upregulated in many types of cancer.5 ,8 ,9 In contrast, patients with COPD undergo a progressive reduction in total HDAC activity in lung tissue as a result of oxidative stress,7 which is associated with proteasomal degradation of many HDACs.10 HDACs are classified into four groups: class I (HDAC1, HDAC2, HDAC3 and HDAC8) are located primarily in the nuclei; class II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10) are located in the cytoplasm but can shuttle to the nucleus; class III are NAD+-dependent enzymes and known as the sirtuins; class IV is represented by HDAC11 only.9 Class I, II and IV HDACs are Zn2+-dependent enzymes, and can thus be efficiently inhibited by chelating agents such as the hydroxamic acids vorinostat (SAHA) and panobinostat (LBH589).9 ,11 ,12 Importantly, these HDAC inhibitors (HDACi) have been reported as successful anticancer agents as they induce cell cycle arrest and apoptosis in cancer cells by increasing the acetylation status of chromatin and other non-histone proteins.9 ,11 ,12
Despite some reports about the involvement of altered HDAC activity in fibrotic lung fibroblasts and IPF,13–15 regulation and regional expression of HDACs have not been characterised in IPF. This study provides a comprehensive description of class I and class II HDAC expression patterns in the IPF lung. In addition, we can demonstrate that the profibrotic and ‘malignant’ phenotype of IPF fibroblasts can be greatly corrected by incubation with the pan-HDACi LBH589.
Materials and methods
Human lung tissue
Lung tissue samples were obtained from 26 patients with sporadic IPF (mean age±SD: 55.85±8.75 years; 5 females, 21 males), and 16 non-diseased control subjects (organ donors; mean age±SD: 48.92±15.84 years; 7 females, 8 males, 1 unknown). Explanted lungs were obtained from the Department of Thoracic Surgery, Vienna (W. Klepetko). All IPF diagnoses were made according to the American Thoracic Society/European Respiratory Society consensus criteria.1 Four out of 26 patients with IPF were former smokers, 14 were never-smokers and from 8 patients the smoking status is unknown. With regard to control subjects, 3 out of 16 were former smokers, 7 were never-smokers and from 6 controls the smoking status is unknown.
Isolation of primary human lung fibroblasts
Primary human lung fibroblasts were isolated from explanted IPF (n=8) and control lungs (n=6) using an outgrowth technique as published.3 Experiments were carried out with IPF/control fibroblasts between passages 3 and 4.
Cell culture experiments
At 95–99% confluency, IPF fibroblasts (n=6) were incubated for 30 h with the HDACi LBH589 (panobinostat, 85nM, Selleckchem) and valproic acid (VPA, 1.5 mM, Santa Cruz), and as control experiment with the respective solvents (vehicle control) in the concentration 0.1% ethanol and 0.03% dimethyl sulfoxide. The dosages of HDACi were chosen according to published12 ,16 and own preliminary studies.
Methodology
Full details for quantitative immunoblotting, immunohistochemistry (IHC), semiquantitative RT-PCR and qRT-PCR (including primers) and statistics are available in the online supplementary material.
Results
Upregulation of class I and class II HDACs in FF and abnormal bronchiolar epithelium in IPF lungs
Comparative immunoblot analysis of subpleural lung tissues from 26 patients with IPF versus 16 non-diseased control lungs revealed a significant upregulation of the class I HDACs HDAC1, HDAC2, HDAC3 and HDAC8 (see online supplementary figures S1A–D), as well as of the class IIa HDACs HDAC4, HDAC5, HDAC7 and HDAC9 (see online supplementary figures S2A–E), which comprises several isoforms (see online supplementary figures S2D, E). Protein expression of the class IIb HDAC10 was insignificantly upregulated (see online supplementary figure S2F). Though class IIb HDAC6 protein expression was not detectable by western blot, qRT-PCR indicated slightly increased HDAC6 mRNA levels in subpleural IPF lung tissues compared with controls (see online supplementary figure S3).
Next, we employed IHC in order to identify the cellular distribution of these enzymes. A nuclear immunostaining for HDAC1 was observed in myofibroblasts of FF, which were identified by staining for alpha smooth muscle actin (α-SMA) (figure 1A, dashed arrows). Prominent nuclear expression of HDAC1 was also observed in overlying, hyperplastic bronchiolar basal cells (figure 1A, indicated by hashmark and cytoplasmic cytokeratin-5/KRT5 staining). Likewise, strong nuclear induction in FF and the overlying bronchiolar epithelium was encountered for the other class I members HDAC2/HDAC3 (see figure 1B and supplementary figures S4A/C). In addition, basal cell sheets of abnormal hyperplastic bronchioles in areas of bronchiolisation (indicated by KRT5 and nuclear p63 staining) showed strong nuclear expression of HDAC1 (figure 1C) and HDAC2/ HDAC3 (see online supplementary figure S5). HDAC2 and HDAC3 also revealed robust nuclear overexpression in ciliated bronchial cells in abnormal bronchiolar structures of IPF lungs (see online supplementary figures S4B and S6). Of note, neither HDAC1, HDAC2, nor HDAC3 showed a significant staining in type II alveolar epithelial cells (AECII) of IPF lungs (figure 1C, D, AECII indicated by arrows and proSP-C staining). In control lungs, HDAC1, HDAC2 and HDAC 3 expression was faint or nearly absent in AECII (figure 1E, indicated by arrows), and only weak expression of HDAC1, HDAC2 and HDAC3 could be encountered in basal and ciliated bronchial cells of normal bronchioles (see figure 1F and online supplementary figure S7).

Induction of class I histone deacetylases in fibroblast foci (FF) and abnormal bronchiolar epithelium of idiopathic pulmonary fibrosis (IPF) lungs. (A) Representative immunohistochemistry for cytokeratin-5 (KRT5), histone deacetylase 1 (HDAC1) and alpha-smooth muscle actin (α-SMA) in serial sections of IPF lung tissue. Strong nuclear staining for HDAC1 was predominantly observed in myofibroblasts of FF (indicated by dashed arrows and α-SMA-staining of a serial section) as well as in overlying hyperplastic bronchiolar basal cells (indicated by hashmark and KRT5 staining) of IPF lungs. (B) Representative immunohistochemistry for HDAC2, HDAC3 and KRT5 in serial sections of IPF lung tissue. In IPF, FF as well as overlying abnormal bronchiolar basal cells (indicated by hashmark and KRT5 expression) revealed strong nuclear induction of HDAC2 and HDAC3. (C) Representative immunohistochemistry for KRT5, HDAC1, prosurfactant protein C (proSP-C) and p63 in serial sections of IPF lung tissue. In IPF, bronchiolar basal cell sheets of hyperplastic bronchioles (positive for KRT5 and p63) indicated robust nuclear induction of HDAC1, whereas type II alveolar epithelial cells (AECII, indicated by arrows) indicated no expression of HDAC1. Only in very rare cases an expression of HDAC1 was observed in the IPF-AECII. AECII are identified by proSP-C immunostaining. (D) Representative immunohistochemistry for proSP-C, HDAC2, HDAC3 and KRT5 in serial sections of IPF lung tissue. IPF-AECII (indicated by arrows and proSP-C staining) did neither express HDAC2 nor HDAC3, which were found in the nucleus of bronchiolar basal cells in abnormal epithelia (positive for KRT5) as well as in some interstitial inflammatory cells (indicated by asterisk) in the very same IPF lung section. (E) Representative immunohistochemistry for proSP-C, HDAC1, HDAC2 and HDAC3 in serial sections of control lung tissue. No or only faint staining for HDAC1, HDAC2 and HDAC3 was observed in AECII of normal lungs; interstitial inflammatory cells (indicated by asterisk) of control lungs showed nuclear HDAC3 expression. (F) Representative immunohistochemistry for HDAC1 and KRT5 in serial sections of control lung tissue. Basal cells of normal bronchioles in control lungs (indicated by KRT5 staining) revealed no notable HDAC1 expression.
Interestingly, robust cytoplasmic expression of HDAC3, but not of HDAC2, was detected in non-ciliated Clara cells of IPF and control lungs (see online supplementary figures S6 and S7B). The same observation was made in IHC for class I member HDAC8, which was expressed in general by bronchiolar epithelium in IPF and control lungs including basal, non-ciliated and ciliated bronchial cells as well as by vascular smooth muscle cells (see online supplementary figures S8B, C). Moreover, cytoplasmic overexpression of HDAC8 was observed in myofibroblasts of FF (see online supplementary figure S8A), which is in line with previous reports describing that cytoplasmic HDAC8 associates with α-SMA and acts as an essential regulator of smooth muscle cell contractility.17
Concerning the cellular distribution of class II HDACs, HDAC4 was abundantly expressed by myofibroblasts of FF (figure 2A, dashed arrows) and in the overlying hyperplastic bronchiolar epithelium (marked by KRT5 staining, figure 2A), as well as in basal cell sheets of abnormal bronchiolar structures in IPF lungs (see online supplementary figure S9A), thereby indicating predominantly cytoplasmic localisation. A similar expression pattern was observed for class IIb HDAC10 (figure 2A, dashed arrows). Furthermore, HDAC4 was robustly expressed in the mature smooth muscle in areas of established fibrosis (indicated by α-SMA staining in figure 2B and online supplementary figure S10B), and co-localised with nuclear HDAC2 and survivin expression (figure 2B). Interestingly, ciliated bronchial cells of IPF lungs indicated strong nuclear expression of HDAC4 (indicated by arrowheads and FOXJ1 staining, figure 2C and supplementary figures S9B and S10C), which appeared to be weak in ciliated epithelium of normal bronchioles in control lungs (figure 2D). Notably, IPF-AECII did not indicate a significant expression of HDAC4 (figure 2C, arrows), whereas robust nuclear expression of HDAC4 was detected in the AECII of control lungs (figure 2D, arrows, and online supplementary figure S11A).

Overexpression of class II histone deacetylases (HDACs) in fibroblast foci and abnormal bronchiolar epithelium of idiopathic pulmonary fibrosis (IPF) lungs. (A) Representative immunohistochemistry for p63, KRT5, HDAC4, alpha smooth muscle actin (α-SMA) and HDAC10 in serial sections of IPF lung tissue. In IPF, immunostaining for HDAC4 (cytoplasmic) and HDAC10 (nuclear and cytoplasmic) was observed in myofibroblasts of fibroblast foci (FF) (indicated by dashed arrows and α-SMA staining of serial sections) as well as in overlying hyperplastic bronchiolar basal cells (indicated by hashmarks, positive for p63 and KRT5). (B) Representative immunohistochemistry for KRT5, HDAC2, HDAC4, α-SMA and survivin in serial sections of IPF lung tissue. Established smooth muscle (indicated by α-SMA staining) in areas of dense ‘older’ fibrosis in IPF revealed cytoplasmic expression of HDAC4, with concomitant nuclear expression of HDAC2 and survivin. (C) Representative immunohistochemistry for KRT5, proSP-C, HDAC4, Forkhead box protein J1 (FOXJ1) and Clara cell protein 10 (CC10) in serial sections of IPF lung tissue. In IPF, strong nuclear staining for HDAC4 is observed in ciliated bronchial cells (indicated by arrowheads and FOXJ1 staining) and basal cells (positive for KRT5) of abnormal hyperplastic bronchioles. Type II alveolar epithelial cells (AECII, indicated by arrows and proSP-C staining) and Clara cells (positive for CC10) in IPF lungs did not express HDAC4. (D) Representative immunohistochemistry for proSP-C, HDAC4 and KRT5 in serial sections of control lung tissue. In the normal lung, HDAC4 was found to be expressed in the nucleus of AECII (indicated by arrows and proSP-C-staining), and basal cytoplasmic expression of HDAC4 was observed in bronchioles of control lungs. (E) Representative immunohistochemistry for p63, KRT5, HDAC7 and α-SMA in serial sections of IPF lung tissue. In IPF, the antibody for HDAC7 revealed strong cytoplasmic staining of myofibroblasts in FF as well as in overlying hyperplastic bronchiolar basal cells (indicated by hashmarks, positive for p63 and KRT5). (F) Representative immunohistochemistry for proSP-C and HDAC7 in serial sections of IPF lung tissue. AECII in areas of dense fibrotic tissue (indicated by arrows and proSP-C-staining) in IPF showed no or only faint immunostaining for HDAC7, whereas bronchial epithelium (BE) indicated robust cytoplasmic HDAC7 expression.
With regard to other class IIa HDAC members, HDAC7 immunostaining was strikingly pronounced in FF and bronchiolar epithelial cells of IPF lungs (figure 2E, F), but was faint in both IPF and donor AECIIs (see figure 2F, arrows, and online supplementary figure S11B). As expected, the expression of the other class IIa HDACs HDAC5 and HDAC9 isoform histone deacetylase-related protein (HDRP) was similarly distributed in IPF and control lungs (see online supplementary figures S12–S18).
Next, cellular localisation of class IIb member HDAC6 was characterised in IPF and control lungs. HDAC6, which has been reported to function in aggresome-autophagy pathway,18 was the only HDAC with pronounced expression in AECII of IPF lungs. Moreover, HDAC6 cytoplasmic expression was upregulated in IPF-AECII compared with control-AECIIs (see online supplementary figures S19A,C, indicated by arrows). In addition, HDAC6 expression was increased in proSP-C-negative, AECII-like (or AECI-like) cells (see online supplementary figure S20). Compared to HDAC7 expression, HDAC6 expression in myofibroblasts was mild (see online supplementary figure S19B). Notably, the ciliated epithelium in IPF and control lungs exhibited abundant cytoplasmic expression of HDAC6 (indicated by hashmarks in online supplementary figures S19A–C).
The IHC expression patterns for all described HDAC enzymes are summarised in table 1.
IHC expression pattern of HDAC enzymes in IPF and control lungs
HDAC expression is upregulated in primary IPF fibroblasts
In accordance with previous reports showing persistence of a profibrotic phenotype of IPF-originating fibroblasts through several passages, we were able to detect a robust and significant upregulation of transcripts for the extracellular matrix (ECM)-associated genes COL1A1, COL3A1 and P4HTM in IPF (n=8) versus control (n=6) fibroblasts (see online supplementary figures S22A–D). ACTA2 appeared only slightly upregulated on mRNA level in IPF (see online supplementary figures S22A/E). In line with mRNA expression, protein biosynthesis of procollagen I was significantly upregulated in IPF versus control fibroblasts (see online supplementary figure S22F). Moreover, primary IPF fibroblasts indicated a significant downregulation of CD90/Thy-1 antigen versus non-diseased controls (see online supplementary figure S22G), which is in line with many previous studies reporting loss of this cell surface glycoprotein in fibroblast populations of IPF lungs, especially in the myofibroblasts of FF.19
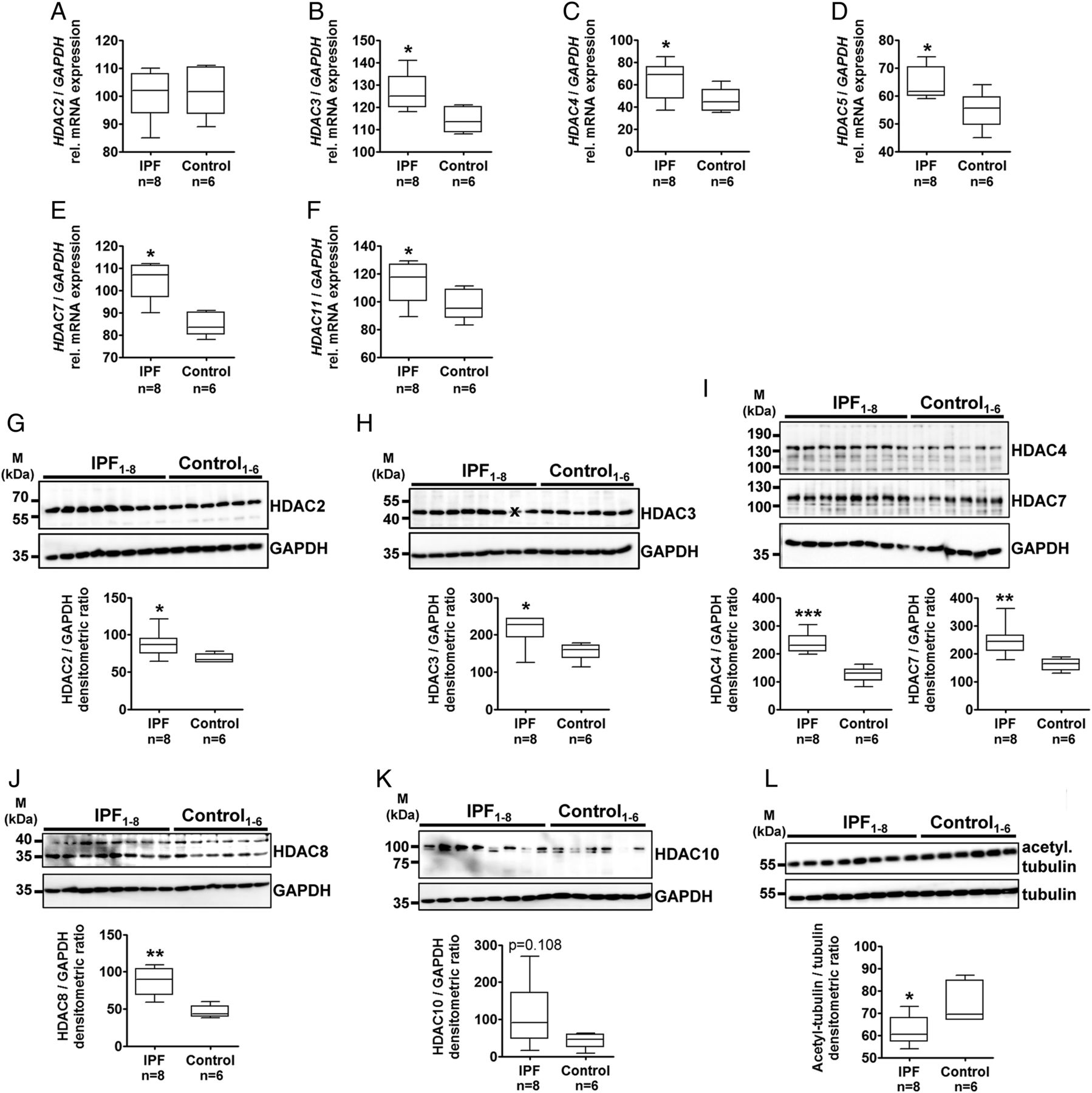
Furthermore, RT-PCR indicated similar expression of HDAC2 mRNA in IPF and control fibroblasts (figure 3A), but significant upregulation of mRNA transcripts for HDAC3 (figure 3B), HDAC4 (figure 3C), HDAC5 (figure 3D), HDAC7 (figure 3E) and HDAC11 (figure 3F) in IPF fibroblasts. As anticipated by IHC analysis, quantitative immunoblotting revealed significant elevated protein expression of HDAC2 (figure 3G), HDAC3 (figure 3H), HDAC4 and HDAC7 (figure 3I), HDAC8 (figure 3J) and HDAC10 (figure 3K) in IPF versus control fibroblasts. HDAC9 protein expression was equal in IPF and control fibroblasts (see online supplementary figure S24). Additionally, IPF fibroblasts indicated reduced tubulin acetylation, which is indicative of increased expression of class IIb HDAC6 and HDAC10 (figure 3L).

Expression analysis of histone deacetylases (HDACs) in primary fibroblasts isolated from idiopathic pulmonary fibrosis (IPF)–and non-diseased control lungs. (A–F) Reverse transcription-PCRs (RT-PCR) for HDAC2 (A), HDAC3 (B), HDAC4 (C), HDAC5 (D), HDAC7 (E) and HDAC11 (F) of human fibroblasts isolated from peripheral explanted lung tissue of patients with IPF (n=8) and non-diseased control lungs (control, n=6). Each PCR reaction was performed with 100 ng reverse-transcribed complementary DNA, followed by electrophoresis through a 2% (w/v) agarose gel containing ethidium bromide. Scanned agarose gels of indicated genes are shown in online supplementary figure S23. Band intensities of PCR products were densitometrically quantified, and mRNA expression of HDAC genes was normalised to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). *p<0.05. (G–L) Representative immunoblots and quantitative immunoblot analysis of human fibroblasts isolated from peripheral lung tissue of patients with sporadic IPF (n=8) and non-diseased control lungs (control, n=6) for HDAC2 (G), HDAC3 (H), HDAC4 and HDAC7 (I), HDAC8 (J), HDAC10 (K), acetylated tubulin (L) and GAPDH or tubulin as loading controls. *p<0.05, **p<0.01, ***p<0.001. Densitometric ratios of the respective gene to GAPDH, as well as of the respective protein to GAPDH or tubulin, are depicted as a box-and-whisker diagram (box indicates 25th and 75th, horizontal line indicates the 50th percentile (median), and extensions above and below reflect extreme values); statistics were performed by Mann–Whitney test. x indicates this sample was not included for statistical analysis due to artefact.
Impact of HDACi treatment on survival and ECM production of IPF fibroblasts
Primary IPF fibroblasts (n=6) were treated with the pan-HDACi LBH589 (85nM) or the class I HDACi VPA (1.5 mM) or vehicle (Veh.) for 30 h, and analysed for cellular consequences. In LBH589 versus vehicle-treated IPF fibroblasts, mRNA expression of the matrix components ACTA2 (figure 4A), COL1A1 (figure 4B), COL3A1 (figure 4C) and FN (figure 4D) were significantly decreased. Likewise, expression of the proliferation marker CCND1 (figure 4E) and the survival gene BIRC5 (figure 4F) was reduced, whereas mRNA levels for the proapoptotic genes CIP1 (figure 4G) and PUMA (figure 4I) were increased. Notably, significant impairment of survival signalling was also observed in VPA-treated cells (figure 4E–G, I). However, a significant reduction of P53 mRNA in response to LBH589 or VPA treatment (figure 4H) was observed, despite increases of p53 target genes CIP1 and PUMA in both conditions. In line with apoptotic signalling, significant induction of the endoplasmic reticulum (ER) stress-associated proapoptotic transcription factor CHOP was observed in LBH589 versus vehicle-treated cells, but not in VPA treatments (figure 4K). The mRNA expression of the ER stress marker ATF6 was slightly elevated in response to LBH589 treatment (figure 4J). Finally, VPA treatment leads to elevated transcription of the E2 conjugase UBE2L6 (figure 4L) that has been described as part of the ubiquitination machinery for HDAC2, which is selectively degraded by the proteasome in response to VPA.16

Cellular signalling in primary idiopathic pulmonary fibrosis (IPF) fibroblasts in response to treatment with panobinostat or valproic acid (VPA). Primary IPF fibroblasts (n=6) were incubated for 30 h with vehicle (0.03% dimethyl sulfoxide, 0.1% ethanol), panobinostat (LBH589, 85 nmol) or VPA (1.5 mM). The effects of vehicle and histone deacetylase (HDACs) inhibitor treatments were analysed by semiquantitative reverse transcription-PCR (RT-PCR) for indicated genes. (A) ACTA2, (B) COL1A1, (C) COL3A1, (D) FN, (E) CCND1, (F) BIRC5, (G) CIP1, (H) P53, (I) PUMA, (J) ATF6, (K) CHOP and (L) UBE2L6. Each PCR reaction was performed with 100 ng reverse-transcribed complementary DNA, followed by electrophoresis through a 2% (w/v) agarose gel containing ethidium bromide. Scanned agarose gels of indicated genes are shown in online supplementary figure S25. Band intensities of PCR products were densitometrically quantified, and mRNA expression of indicated genes was normalised to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are presented as mean±SEM of the individual values of different treatments. *p<0.05, **p<0.01, ***p<0.001, LBH589 or VPA vs vehicle; p<0.05, p<0.01, p<0.001 VPA vs LBH589; by Dunn's multiple comparison test.
Next, gene expression of HDAC enzymes in response to HDACi treatment was analysed (see online supplementary figures S26A–N). Compared to vehicle-treated cells, LBH589 treatment resulted in reduced HDAC11 expression (see online supplementary figure S9L) and to marked suppression of HDAC7 (see online supplementary figure S9H), paralleled by a significant increase in HDAC3 (see online supplementary figure S9D) and SIRT2 transcripts (see online supplementary figureS9N).
On protein level (see figure 5 and online supplementary figure S27), IPF fibroblast lysates (n=4) indicated a significant increase of total acetylated histone H3 after treatment with LBH589 or VPA versus vehicle (figure 5A), whereas significant tubulin-acetylation was detected only in LBH589-treated cells (figure 5B) due to efficient inhibition of class IIb HDAC6 and HDAC10. In line with mRNA expression analysis, p21 protein was significantly increased (despite reduction of p53) in LBH589-treated IPF fibroblasts compared with vehicle control (figure 5C, D). Furthermore, western blot analyses displayed a decrease in proliferation marker phospho-histone H3 in LBH589-treated as well as VPA-treated IPF fibroblasts (figure 5E). Cyclin D1 was reduced only in LBH589-treated cells (figure 5F). Concomitantly, antiapoptotic proteins survivin (figure 5G) and Bcl-XL (figure 5H) were reduced, with parallel induction of the proapoptotic ER stress marker CHOP (figure 5I) and caspase-3 activation (figure 5J), in LBH589-treated, but not VPA-treated IPF fibroblasts. It has been already shown in cancer cells that ER stress plays a pivotal role in cell death mediated by LBH589, independent of p53.20

Analysis of acetylation status, apoptotic signalling and profibrotic protein expression in primary idiopathic pulmonary fibrosis (IPF) fibroblasts in response to treatment with panobinostat or valproic acid (VPA). Primary IPF fibroblasts (n=4) were incubated for 30 h with vehicle (0.03% dimethyl sulfoxide, 0.1% ethanol), panobinostat (LBH589, 85 nmol) or VPA (1.5 mM). Status of acetylation, apoptosis, histone deacetylase (HDAC) protein expression and profibrotic protein expression was analysed by quantitative immunoblotting. Representative immunoblots of indicated research targets are shown in online supplementary figure S27. In dependency of research target, histone H3, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-actin or tubulin served as loading control. (A) Histone H3-acetyl K27, (B) acetylated tubulin, (C) p53, (D) p21, (E) phospho-histone H3, (F) cyclin-D1, (G) survivin, (H) Bcl-Xl, (I) CHOP, (J) caspase-3, (K) HDAC7, (L) HDAC2, (M) HDAC3, (N) alpha-smooth muscle actin (α-SMA), (O) COL1A1 (pro-form) and (P) COL1A1 (mature form). Data are presented as mean±SEM of the individual values of different treatments. *p<0.05, **p<0.01, ***p<0.001, LBH589 or VPA vs vehicle; p<0.05, p<0.01, p<0.001 VPA vs LBH589; by Dunn's multiple comparison test.
In accordance with suppression of HDAC7 gene expression in response to LBH589 treatment, protein expression of HDAC7 was almost absent under these conditions, but also reduced in VPA-treated IPF fibroblasts in comparison to vehicle control (see figure 5K and online supplementary figure S28; two different antibodies were tested for HDAC7 expression). In line with this interesting observation, the pan-HDACi SAHA has been shown to selectively suppress expression of HDAC7 in several cell lines.21
Furthermore, VPA, but not LBH589 treatment, resulted in a significant downregulation of HDAC2 protein expression (figure 5L), presumably through E2-conjugase UBE2L6-mediated polyubiquitination and proteasomal degradation of HDAC2, a process that has been observed in several cell lines in response to VPA.16 In contrast, protein expression of HDAC3 was not altered by VPA, but was significantly upregulated in LBH589-treated versus vehicle-treated IPF fibroblasts (figure 5M).
Finally, all these changes were accompanied by significant reduction of collagen I biosynthesis in both LBH589-treated and VPA-treated IPF fibroblasts versus vehicle treatment, as assessed by immunoblotting for pro-forms (figure 5O) and mature forms of collagen I (figure 5P). Of note, VPA treatment did not reduce α-SMA expression in IPF fibroblasts (figure 5N).
Discussion
The development, growth and aggressiveness of cancer cells have been associated with upregulated HDAC expression. Class I HDACs such as HDAC2 appear to be crucial for the regulation of proliferation and apoptosis resistance of cancer cells, in part through their ability to inhibit the tumour suppressor p53 and to promote expression of the proto-oncogene c-Myc.8 Several publications have also implicated many roles of class II HDACs in tumourigenesis: HDAC4 promotes growth of colon cancer cells via repression of p21.22 HDAC5 strongly interacts with the tumour suppressor RUNX3 and induces its degradation through deacetylation.23 HDAC7 has been shown to protect from apoptosis by inhibiting c-Jun expression24 and contributes to carcinogenesis by transcriptional activation of c-Myc.25 However, little was known about expression and function of HDACs in IPF. In this paper, we show for the first time that nearly all class I and II HDAC enzymes are overexpressed and upregulated in IPF lung tissue. In detail, upregulation of HDACs was predominantly observed in myofibroblasts of FF as well as in abnormal bronchiolar basal cells in areas of bronchiolisation in IPF lungs, and co-localised with expression of survivin. These results are not surprising as key features in the pathogenesis of IPF include expansion of the population of fibroblasts and their differentiation into apoptosis-resistant myofibroblasts, which consecutively produce large amounts of ECM components.1 While class I HDACs revealed strong nuclear induction in FF, class IIa HDACs HDAC4, HDAC5, HDAC7 and HDAC9 were predominantly expressed in the cytoplasm of myofibroblasts. The translocation of class IIa HDACs from the nucleus to the cytoplasm is associated with activation of transcription factors myocyte-enhancer factor-2 and serum response factor, which activate myogenic genes such as ACTA2, and which are repressed by nuclear localisation of class IIa HDACs.14 Additional studies using RNAi technology identified HDAC4 as an HDAC that mediates transforming growth factor (TGF)-β1-induced differentiation of normal lung fibroblasts into myofibroblasts. Also here, the cytoplasmic localisation of HDAC4 was required for activation of AKT signalling pathway, which is necessary for ACTA2 expression in response to TGF-β1.14 We suggest that ‘cancer-like’ upregulation of class I and II HDACs in IPF fibroblasts is associated with (i) increased fibroblast proliferation, (ii) fibroblast-to-myofibroblast differentiation and (iii) with the apoptosis-resistant phenotype of myofibroblasts. Another striking hallmark of IPF is the so-called ‘bronchiolisation-process’ that describes the aberrant progressive re-epithelialisation of damaged alveolar epithelium by proliferating bronchiolar basal cells.1 ,26 Based on our data, we suggest that the exaggerated, proliferative character of basal cells in IPF may be in part due to overexpression of HDACs and that HDACs may govern the process of aberrant bronchiolisation in this disease. In line with this, luminal ciliated bronchial cells (which stem from the progenitor basal cells) indicated also robust overexpression of class I (especially HDAC2 and HDAC3) and of all class II HDACs, whereas only faint or moderate expression was observed in the ciliated epithelium of non-diseased control lungs. Moreover, basal and luminal epithelial cells near areas of active fibrotic remodelling and aberrant bronchiolar proliferation or of abnormal IPF bronchioles that often appear hyperplastic and larger in size are suspected to contribute to the abnormal, exaggerated production of profibrotic signals in IPF26 and that thus may require enhanced overexpression of ‘cancer-like’ HDACs. Of note, expression of many HDAC enzymes seemed to be virtually absent or was only faint in normal bronchiolar structures as well as AECII (or rather in the whole lung tissue) of non-diseased control lungs, presumably due to their crucial intrinsic activity in cell proliferation and apoptosis inhibition,5 ,8 ,9 which justifies an only low and basal expression in normal, healthy cells.
The class IIa HDACs HDAC4 and HDAC7 were the only HDACs with moderate to robust expression in ciliated epithelium and AECII of non-diseased control lungs. However, we can only speculate about the function of both these HDACs in the normal AECII. HDAC4 and HDAC7 have been both reported to regulate the functional activity of HIF-1α in various cell types in vitro because loss of each of them resulted in reduced HIF-1α transcriptional activity of adaptive responses to hypoxia.27 ,28 For alveolar-epithelial expressed HIF-1α, a surprising role in lung protection during acute lung injury (ALI) has recently been reported in which normoxic HIF-1α stabilisation and HIF-1α-dependent control of alveolar-epithelial glucose metabolism attenuated lung inflammation during ALI induced by stretch conditions in vitro or by ventilator-induced lung injury in vivo.29 Additionally, HIF-1α has been reported to be essential for proper lung maturation and surfactant production in the newborn lung of mice.30 Based on these studies, we can presume that HDAC4 and HDAC7 might play a crucial role in maintaining AECII homeostasis through regulation of HIF-1α.
Though, and in marked contrast to bronchiolar epithelial cells in IPF, AECII of IPF lungs did not reveal significant expression of class I and II HDACs (which were in part significantly expressed in AECII of normal lungs, such as HDAC4 and HDAC7 as described above), possibly due to pro-apoptotic ER and oxidative stress in IPF-AECII,31–33 which in turn lead to proteasomal degradation of many HDACs. Moreover, HDAC4 is a death substrate, cleaved by caspase-3 during apoptotic cell death,34 and HDAC7 is the best caspase-8 substrate described to date.35
Importantly, the class IIb member HDAC6 was the only HDAC expressed in IPF-AECII, but not in AECII of normal lungs. As a tubulin deacetylase, HDAC6 is involved in aggresome formation and autophagic clearance of protein aggregates in response to increased misfolding,18 and its induction in AECII can be due to severe alveolar epithelial ER stress in IPF.31–33 Additionally, HDAC6 deacetylates antioxidants PRDX1 and PRDX2, which impairs their function to reduce H2O2, thereby promoting apoptosis.36 Despite its reported pro-apoptotic and pathological function, it seemed to be an intrinsic normality of ciliated bronchial cells of IPF as well as non-diseased control lungs to robustly express HDAC6 in the cytoplasm, as shown by our IHC studies. Of note, autophagy is also critical for cell survival because unwanted intracellular materials are degraded, and HDAC6 controls autophagosome maturation through its deacetylase activity.37 Indeed, it was shown that cells deficient in HDAC6 failed to form autophagosomes and to clear misfolded protein aggregates, and were hypersensitive to the accumulation of malfolded proteins.37 On the other hand, inhibition of HDAC6 deacetylase activity by pan-HDACi such as SAHA or trichostatin-A has been shown to ameliorate disease progression and neurodegeneration in rodent models of Huntington’s disease and amyotrophic lateral sclerosis.38 ,39 Anyway, the exact role of HDAC6 in the ciliated lung epithelium and the impact of its induction in IPF-AECII remain still elusive and have to be further investigated.
More than 10 HDACi have already been examined in clinical trials as cancer drugs and only minor side effects have been observed.9 HDACi possess the ability to selectively induce apoptosis in tumour cells, whereas normal cells are relatively resistant to HDACi-induced cell death.9 ,11 ,40 In addition to antitumour activities, HDACi have been shown to exert beneficial effects on several diseases such as cardiac hypertrophy,6 inflammatory bowel disease41 and above-mentioned neurodegenerative diseases.38 ,39 The present study suggests that HDACi such as LBH589 may also be useful for the treatment of IPF. Interfering with the generation of myofibroblast foci to decrease production of ECM components is important in treating IPF because the increase in the number of these fibrotic foci has been associated with disease progression and a worsened prognosis.42 We demonstrated that LBH589 downregulated significantly the mRNA expression of ACTA2 and ECM genes COL1A1, COL3A1 and FN in primary IPF fibroblasts and thus interfered with fibroblast-to-myofibroblast differentiation. In addition, LBH589 efficiently reduced the protein biosynthesis of the large collagen α-1 chains (COL1A1) in IPF fibroblasts. The reduction in ECM synthesis was accompanied by growth inhibition as shown by immunoblotting for p-histone H3 and cyclin-D1. In parallel, LBH589 suppressed expression of the survival-related HDAC7 gene and downregulated expression of antiapoptotic genes BCL2L1/Bcl-XL and BIRC5/survivin in IPF fibroblasts, and led to induction of ER stress and apoptosis, thereby resulting in efficient inactivation of ECM producing and ‘malignant appearing’ IPF fibroblasts. Moreover, in renal fibrosis, it was demonstrated in a murine model of unilateral ureteral obstruction that trichostatin-A led simultaneously to inactivation of renal interstitial fibroblasts and inhibition of renal tubular epithelial cell death,43 thus adding further experimental proof to use pan-HDACi for the treatment of IPF. Furthermore, it should be noted that the treatment of IPF fibroblasts with VPA, which has been described as a weak class I HDACi, led also to a significant reduction in collagen I biosynthesis. Additionally, VPA treatment induced significant downregulation of HDAC2 protein levels in IPF fibroblasts, presumably through UBE2L6-mediated polyubiquitination and proteasomal degradation of HDAC2.16 Because overexpression of HDAC2 is a classical ‘cancer condition’, drugs that interfere with HDAC2 protein turnover in myofibroblasts are of great importance.
The exact targets of HDACs in the setting of fibrosis are yet not clear. Similar to cancer cells, in which ‘aberrant transcriptional repression’ has been described as a result of class I HDAC overexpression,5 ,8 ,11 ,16 we suggest that target genes of class I HDACs in lung fibroblasts might be repressors of antiapoptotic and profibrotic gene expression (which are silenced in the setting of fibrosis due to class I HDAC upregulation). The contribution of class IIa HDACs to lung fibrosis is presumably antiapoptosis22–25 as well as maintaining of cytoplasmic AKT phosphorylation/activation and consequent promotion of profibrotic gene expression.14
In conclusion, based on the results of our study, HDACs are novel molecular targets for IPF therapy, and pan-HDACi such as LBH589 are promising therapeutic agents for the treatment of IPF, even though their exact targets and mechanisms of action in the setting of IPF must be further investigated. The main observations and suggestions from this study are summarised in figure 6.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Aberrant histone deacetylase (HDAC) overexpression likely contributes to idiopathic pulmonary fibrosis (IPF) pathogenesis. (A) In the healthy lung with normal alveolar architecture, histone acetylation and deacetylation is balanced and contributes to appropriate gene transcription of pivotal cellular signalling pathways. The triggering event for IPF is alveolar epithelial cell (AEC) injury and death. AECII cells become hyperplastic and depleted of HDACs (except for HDAC6); endoplasmic reticulum stress and apoptosis are also present. In parallel, an aberrant upregulation of class I/II HDACs is observed in different fibroblast populations including myofibroblasts and bronchiolar basal cells. The contribution of injured IPF-AECIIs to the malignant HDAC overexpression in fibroblasts and basal cells in IPF remains to be elucidated. As a result of class I HDAC overexpression, we hypothesise that transcription of repressors of fibrosis is reduced in both fibroblast and basal cell populations of IPF lungs, thereby leading to aberrant activation of proliferative, profibrotic and apoptosis-resistant genes. Class II HDACs contribute to profibrotic gene expression by activating and maintaining cytoplasmic AKT phosphorylation. All together, this leads to abnormal fibroblast proliferation and myofibroblast generation. Alveolar spaces are infiltrated by myofibroblasts and activated resident fibroblasts, which begin to form fibroblast foci. Basal cells proliferate and migrate distally. As IPF progresses, alveolar spaces are progressively replaced by fibrotic tissue, smooth muscle and abnormal bronchiolar epithelium (‘bronchiolisation’). (B) In vitro, pan-HDAC inhibition by LBH589/panobinostat efficiently inactivates primary profibrotic fibroblasts and myofibroblasts through downregulation of extracellular matrix (ECM) production on the one side and induction of apoptosis on the other side, and which thus may have a significant benefit for patients with IPF. (C) Legend.
Acknowledgments
We thank Gabriele Dahlem and Susanna Ziegler for isolation of primary fibroblasts from explanted IPF and non-diseased control lungs.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors MK, OHK and AG designed experiments and wrote the manuscript. MK, SS, IH, BAMK, OK, ShS, LF, CR, DvdB, PM, and WK carried out the experimental work. BAMK, SB, BC, SSP and WS served as key advisors and critically read the manuscript. OHK and AG equally contributed to this manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study protocol was approved by the Ethics Committee of the Justus-Liebig-University School of Medicine (no. 31/93, 29/01, and no. 111/08: European IPF Registry).
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves