Article Text

Abstract
Aim Acute bronchiolitis is the commonest cause for hospitalisation in infancy. Supportive care remains the cornerstone of current management and no other therapy has been shown to influence the course of the disease. It has been suggested that adding nebulised hypertonic saline to usual care may shorten the duration of hospitalisation. To determine whether hypertonic saline does have beneficial effects we undertook an open, multi-centre parallel-group, pragmatic RCT in ten UK hospitals.
Methods Infants admitted to hospital with a clinical diagnosis of acute bronchiolitis and requiring oxygen therapy were randomised to receive usual care alone or nebulised 3% hypertonic saline (HS) administered 6-hourly. Randomisation was within 4 h of admission. The primary outcome was time to being assessed as ‘fit’ for discharge with secondary outcomes including time to discharge, incidence of adverse events together with follow up to 28 days assessing patient centred health related outcomes.
Results A total of 317 infants were recruited to the study. 158 infants were randomised to HS (141 analysed) and 159 to standard care (149 analysed). There was no difference between the two arms in time to being declared fit for discharge (hazard ratio: 0−95, 95% CI: 0.75−1.20) nor to actual discharge (hazard ratio: 0.97, 95% CI: 0.76−1.23). There was no difference in adverse events. One infant in the HS group developed bradycardia with desaturation.
Conclusion This study does not support the use of nebulised HS in the treatment of acute bronchiolitis over usual care with minimal handlings.
ClinicalTrials.gov registration number NCT01469845.
- Paediatric Lung Disaese
- Viral infection
- Nebuliser therapy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
Does nebulised hypertonic saline influence the duration of hospitalisation when used to treat infants with acute bronchiolitis?
What is the bottom line?
Nebulised hypertonic saline does not reduce the length of stay in infants hospitalised with acute bronchiolitis.
Why read on?
This definitive multicentre study will directly influence clinical care to prevent the adoption of an ineffective treatment and provide a model for the conduct of future studies involving inhaled therapy in this condition.
Introduction
Acute bronchiolitis is the most common cause for hospitalisation in infancy with 1%–3% of all infants being admitted during their first winter.1–4 The disease is caused by a number of common respiratory viruses, with respiratory syncytial virus (RSV) the most commonly identified, and is associated with the characteristic winter peaks in admissions.1–7 Infants typically develop signs of an upper respiratory tract infection with rhinitis followed by signs of lower respiratory tract involvement due to lower airway obstruction. This is manifest by increased work of breathing, hyperinflation, cough and widespread crackles on auscultation which may or may not be accompanied by wheeze.1–5 The peak age incidence of babies admitted to hospital with this condition is between 1 and 6 months.1–9
Supportive care, with supplemental oxygen to correct hypoxia, minimal handling to minimise the risk of exhaustion and the provision of fluids, remains the cornerstone of management.1–5 ,7–11 The introduction of oxygen therapy, over 60 years ago, reduced mortality rates from as high as 20% in the 1940s11 to substantially <1%,12 ,13 though worldwide it remains a major cause for infant death.14 In the UK, admissions for acute bronchiolitis increased from 21 330 in 2004/2005 to 33 472 in 2010/2011,15 placing enormous strains on paediatric in-patient services and paediatric intensive care units16 which sometimes have to close due to the numbers of infants with acute bronchiolitis. The mean duration for admission in the UK is around 3.3 days,17 compared with a median of 1.5 days for all acute paediatric admissions.18 Since the introduction of oxygen therapy, antiviral agents, oral and inhaled steroids and a variety of bronchodilators have neither decreased lengths of inpatient stay nor impacted on the course of the acute illness,1–4 ,7–10 while an effective vaccine still appears someway off.19
Some studies published over the past 8 years have suggested that nebulised hypertonic saline (HS) may influence the course of the illness and reduce the duration of hospitalisation. A Cochrane review concluded that, ‘nebulized 3% saline may significantly reduce the length of hospital stay and improve the clinical severity score in infants with acute viral bronchiolitis’.20 The studies included in the review were undertaken in a diverse range of healthcare settings with a range of therapies and comparators. HS was used at 3% and 6%, with and without a bronchodilator and compared, in most studies, with nebulised normal saline with and without other agents. Some authors have suggested that normal saline can impact favourably on the clinical outcomes and, as such, is an active comparator,21 while older studies using distilled water as the ‘control’ agent were criticised on the basis that the hypo-osmolar water could induce bronchospasm. A further challenge in interpreting these studies is that the term ‘acute bronchiolitis’ is used to describe phenotypically different patients in different parts of the world. The above definition in which widespread crackles are a characteristic finding is used in the UK, Australia and a number of other countries, while in the USA and other countries it is used to describe the first episode of wheezing with an apparent viral infection.1–4 ,7 While the underlying pathology, dominated by neutrophil influx into the airway,22 may be very similar in most of these patients, the latter definition would include a significant proportion of infants experiencing a first viral exacerbation of asthma.
In light of these challenges, a multicentre, randomised, open, pragmatic study was undertaken in which infants requiring oxygen therapy for acute bronchiolitis were randomised to receive usual care or usual care with 4 mL nebulised 3% HS 6-hourly until they were fit for discharge.
Methods
The carers of all randomised participants provided written informed consent. The study was approved by the South Yorkshire Research Ethics Committee and registered with ClinicalTrials.gov, number NCT01469845.
Study design and participants
Hypertonic Saline in Acute Bronchiolitis Rct and Economic evaluation (SABRE) was a multicentre, parallel-group, open, randomised controlled trial with a 1:1 allocation ratio. An economic evaluation alongside the trial will be reported separately. This report is compliant with CONsolidated Standards of Reporting Trials (CONSORT) 2010.23 Participants were recruited from the assessment units and paediatric wards of 10 participating centres in England and Wales between October 2011 and December 2013.
Eligible participants were healthy infants under 1 year of age needing supplementary oxygen for oxygen saturations of <92% in air when admitted to hospital with a clinical diagnosis of acute bronchiolitis. This was defined as an apparent viral respiratory tract infection associated with airways obstruction manifest by hyperinflation, tachypnoea and subcostal recession with widespread crepitations on auscultation.8 The time of admission was defined as when the paediatrician, or paediatric Advanced Nursing Practitioner, made the decision to admit. Eligible participants required supplemental oxygen therapy on admission and were consented and randomised within 4 h of admission. The following were exclusion criteria: a history of wheezy bronchitis or asthma; gastro-oesophageal reflux; previous lower respiratory tract infections; risk factors for severe disease; carers lacking fluent English in the absence of translational services and patients requiring admission to high dependency or intensive care units (HDU/ICU) at presentation.
Allocation was either to standard supportive care (control) or oxygen as required, and fluid administration plus 4 mL nebulised 3% HS solution (PARI MucoClear) every 6 h, administered by a nurse via the PARI Sprint nebuliser with appropriate face mask (intervention). The nebuliser and face mask were purchased from (PARI Medical, UK). The saline treatment was discontinued once the primary outcome had been achieved. All concomitant medications were recorded. Centres were encouraged to discontinue antibiotics at admission if previously prescribed, but antibiotics were permissible for suspected secondary bacterial infection in line with UK guidance.8 Standard supportive care involved oxygen as required, minimal handling and fluid administration as appropriate to the severity of the disease.
The final protocol with details of amendments are documented elsewhere.24 Initially, patients were to be randomised within 90 min of a decision to admit, but an amendment permitted this to be extended to 240 min given the practical difficulties of meeting the 90 min deadline.
Randomisation and masking
After parental informed consent was obtained, infants were randomised using a centralised web-based randomisation system with a computer generated algorithm generated by Sheffield Clinical Trials Research Unit. Randomisation was conducted in randomly ordered blocks of size two, four and six stratified by hospital. This was an open study when blinding was not possible.
Outcomes
The primary outcome was the time until the infant was assessed as being ‘fit for discharge’, which was defined as the point at which the infant was feeding adequately (taking >75% of their usual intake) and had been in air with a saturation of at least 92% for 6 h. Data was collected on a daily basis from the routinely recorded clinical observations from which the two key outcomes, time fit for discharge and time to actual discharge were calculated. Secondary outcomes included actual time to discharge; readmission within 28 days from randomisation; adverse events; healthcare usage and duration of respiratory symptoms postdischarge. Parents were also asked to complete a validated infant and parental quality of life using the Infant Toddler Quality of Life (ITQoL) questionnaire 28 days after randomisation.25 Data was collected on the ward in a case report form and, after discharge, in a symptom and health service usage diary.
Statistical analysis
Based on data taken from UK hospital episode statistics, we expected an average time to discharge of around 3 days with an SD of 32 h. Assuming a conservative SD of 46 h, in order to have 90% power to detect a 25% difference in time to meeting discharge criteria, the study needed 139 patients per group at a two-sided α level of 5%; this corresponds to an HR of approximately 0.7 on a log-rank test.
The recruitment period was extended to overcome slow recruitment. At the request of the data monitoring committee, we undertook one unplanned interim analysis for efficacy using a 1% significance level after 152 patients were recruited, with the final significance level adjusted downwards to 4.5%.26 The interim analysis report was restricted to the data monitoring committee. All other facets of the analysis remained unchanged from the prespecified analysis plan, which was written prior to data, being available to the statisticians.
The primary outcome was analysed by a Cox proportional hazards regression model in which the centre was fitted as a fixed effect. Proportional hazards was assessed by plotting scaled Schoenfeld residuals against time.27 We also assessed whether HS worked similarly in RSV+ and RSV− patients by including RSV status and its interaction with treatment group.
The proportion of patients admitted to HDU/ICU and the proportion readmitted to hospital were compared between treatment groups by logistic regression, again with centre as a covariate. Each of the nine dimensions of the ITQoL study was analysed by a Mann–Whitney U test. All analyses presented are by intention to treat unless otherwise stated. Analyses were conducted using SPSS V. 20 or SAS V. 9.3.
Role of the funding source
The funder was not involved in the trial design, patient recruitment; data collection, analysis, interpretation, or presentation; writing or editing of the report; or the decision to submit for publication. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
The trial randomised 317 infants between 26 October 2011 and 23 December 2013, when it closed having reached full accrual, with 158 patients allocated to the nebulised 3% HS group and 159 allocated to usual care (figure 1). Twenty-six out of 317 participants were excluded as being ineligible when randomised, and medical notes were incomplete for one eligible participant, leaving 290 participants included in the primary analysis. Baseline demographics are displayed in table 1. In the intention-to-treat (ITT) population, in which patients were analysed by their original assigned groups, there were 142 participants in the HS arm and 149 in the control arm. One intervention group patient did not have any date and time of fit for discharge recorded and are not included in any of these analyses, but were known to be discharged on day 6 (i.e. between 120 h and 144 h); this length of stay (LoS) is similar enough to the remainder for their missing data to have no material impact. Five participants did not receive treatment as scheduled; a sensitivity (per protocol) analysis, in which these five participants were excluded, gave almost identical results as ITT analyses (data not shown).

CONSORT diagram. ITQoL, Infant Toddler Quality of Life; ITT, intention-to-treat.
Demographics
In intent-to-treat analyses (HS n=141; control 149, except where stated), there was no observed difference between the two treatment arms, either in the time to being declared fit for discharge (HR=0.95, 95% CI 0.75% to 1.20%; p=0.66; figure 2) or in the time to actual discharge (HR=0.97, 95% CI 0.76 to 1.23; figure 3 and table 2). The median time to being declared fit for discharge was 76 h from admission in each group, and the time to actual discharge was 89 h in each group. RSV+ status (n=179, median 80.6 h) was associated with a longer time until fitness for discharge than observed in RSV− patients (n=27, median 50.8 h) (HR=1.94, 95% CI 1.24% to 3.02%; p=0.004) and a longer time to actual discharge (medians 91.1 h versus 72.2 h, HR=1.47, 95% CI 0.95% to 2.28%; p=0.09), but there was no evidence of any interaction between treatment effect and viral status (p=0.59 for time to fit for discharge and p=0.41 for time to actual discharge). Viral testing was not undertaken in 79 patients in line with routine practice in some centres.
Time to being declared fit for discharge and time to discharge (hours)

Cumulative survival plot for time to being declared fit for discharge.

Cumulative survival plot for actual time to discharge.
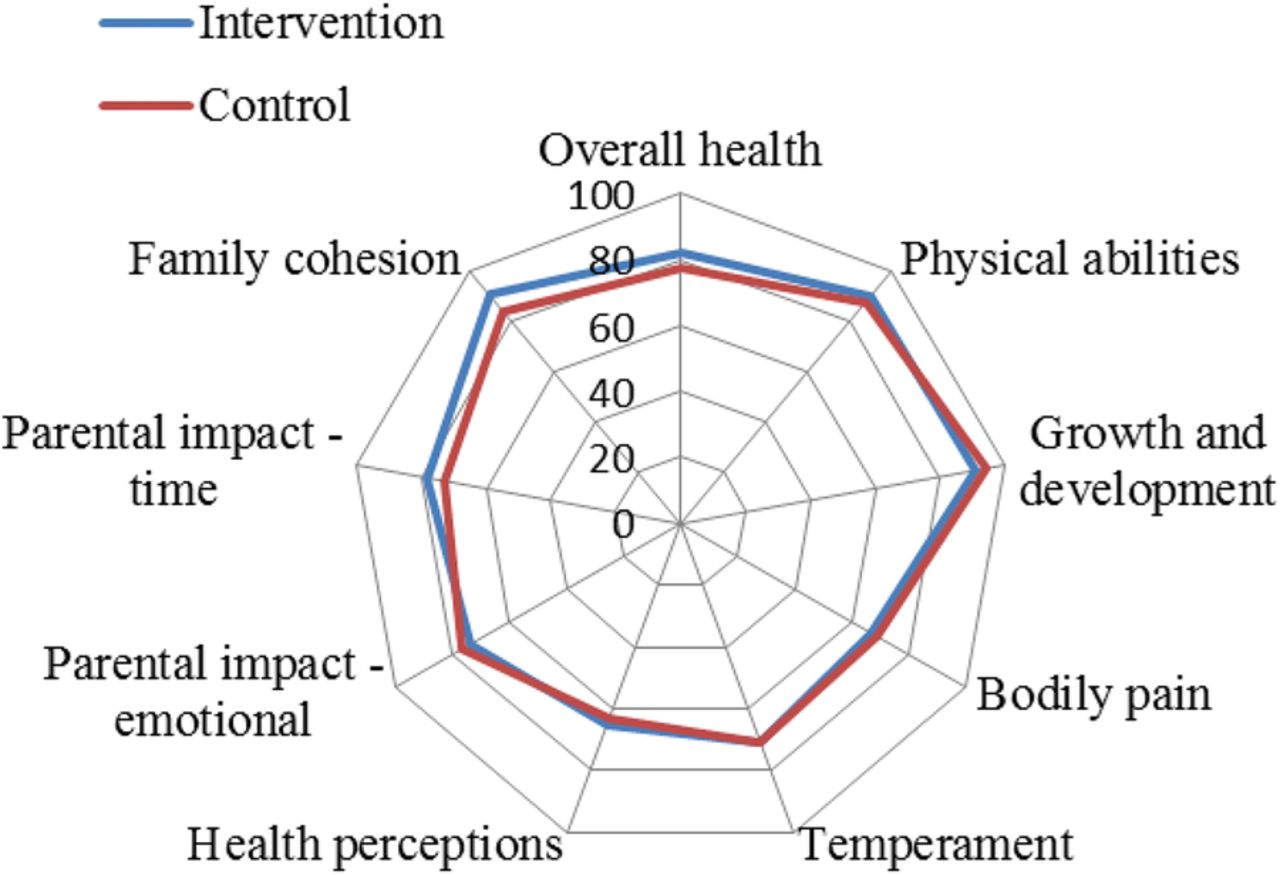
There was no evidence to suggest a difference between treatment groups in terms of the numbers admitted to HDU/ICU or readmitted within 28 days of randomisation (table 3). No important differences were observed on any of the ITQoL dimensions (figure 4: HS n=54; control 49) and there was no evidence that outcomes differed between groups according to RSV status.
Admission to HDU/ICU, readmission rates and symptoms to 28 days
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ITQoL dimensions by study group, full analysis set (FAS). ITQoL, Infant Toddler Quality of Life.
Six adverse events were possibly related to saline treatment, including one serious adverse event (SAE), bradycardia and desaturation during administration of the nebuliser, which resolved the following day. The remaining five non-SAEs were: bradycardia (self-correcting), desaturation, coughing fit and increased respiratory rate (all of which were resolved within 1 day); and a chest infection which resolved after 6 days.
Discussion
The data generated in this randomised study indicate that adding 6-hourly nebulised 3% HS to usual care when treating infants admitted to hospital with acute bronchiolitis has no discernible beneficial or harmful consequences when compared with usual care alone. In particular, there was no change in either the time taken for infants to be assessed as being fit for discharge or in the actual time to discharge. Furthermore, the intervention had no impact on admissions to HDU/ICU, readmission rates, or reported morbidity in the month following the acute illness. Moreover, the conclusion is unaffected by whether the acute illness was caused by the RSV or one of the many other respiratory viruses that can cause the same clinical illness. Consistent with previous studies, 85% of tested infants were positive for RSV.
This is the largest study undertaken to investigate the potential for HS to influence the duration of admission among infants hospitalised with acute bronchiolitis and its results highlight the need for funding large well-designed, randomised, clinical trials in paediatrics. The results are likely to contribute significantly to individual patient care and to resource usage in the healthcare system by preventing the widespread introduction of an ineffective and time-consuming therapy.
Among the strengths of this study was the multicentre design using both teaching hospitals and district general hospitals which ensures that the results are generalisable to all paediatric secondary care units admitting such infants. Moreover, the study was designed to assess the potential role of this intervention in the most severely affected infants, namely infants admitted to hospital requiring supplemental oxygen therapy. Such patients place a significant burden on healthcare systems globally. Nebulised 3% HS was used because this was the concentration used in the majority of previous studies, and because there is a commercial product marketed for this indication. For comparison, the concentration of salt in blood is 0.9% and that in sea water 3.4% (3.2% to 3.8%). There is no indication from the literature that other concentrations of HS would be more effective than 3% saline.
A potential limitation of the study is the absence of blinding. However, we believe that the use of an open pragmatic study design is one of the strengths of the study. This approach was chosen in order to avoid potential confounders and to determine whether the use of nebulised HS might have a place in routine clinical practice. A number of previous studies involving HS and other nebulised interventions, such as the antiviral agent ribavirin, have used a randomised blinded approach using either distilled water or normal saline as the nebulised placebo agent. Both approaches have been criticised in that it is argued that the hypo-osmolar distilled water can induce bronchoconstriction, thus hindering the infant's recovery, while some proponents of hypertonic therapy have argued that failure to show a difference when compared with nebulised normal saline is due to the beneficial effects of the depositing normal saline in the lower airways.21 The remarkable congruity in the outcomes for both arms of the trial, as shown in figures 2 and 3, argue against the influence of any bias resulting from this being a pragmatic study, since any putative therapeutic or adverse effect would have to be exactly matched by the effect of any systematic bias. Moreover, the study was conducted in 10 centres with the key outcomes being obtained by nurses using routinely recorded clinical data. It is extremely unlikely that any systematic view of the potential benefits or harm would influence the many dozens of medical staff involved in the care of these infants.
The study protocol imposed a restrictive time window within which it was permissible to randomise. As a result, we excluded from the study a number of otherwise eligible infants, whom clinicians might consider appropriate candidates for treatment. We have no reason to suppose that infants who entered the trial were substantially different from those who did not, thus biasing the results, but did not collect data on infants without parental consent. The relatively limited data regarding effects of quality of life and other outcomes during the first month after discharge was disappointing, but does not impact on the primary outcome and major secondary outcomes.
There is good evidence that bronchoconstriction does not contribute significantly to the airways obstruction in infants with acute bronchiolitis which explains why bronchodilators do not provide any benefit.1–4 ,7–9 Instead, the obstruction appears to be, in part, due to oedema within the airway wall and, probably more importantly, accumulation of inflammatory exudates in the airways driven by the intense neutrophilia within the lumen of the airways of affected infants.22 Significant impairment of mucocillary clearance due to shedding of ciliated cells, probably, also contributes to accumulation of secretions in the small airways. The suggested mode of action of HS is through an alteration in mucus rheology as a result of improved hydration and the breaking of ionic bonds within the mucus leading to improvements in mucocillary clearance of secretions.21 ,28 The observation that HS can increase ciliary beat frequency may further enhance clearance. It is also suggested that this intervention reduces mucosal wall oedema through osmotic effects.21 ,28 At present, there is no direct in vivo data to support the suggestion that the luminal surface of the infected airways is effectively in a dehydrated state in vivo, specifically in RVS bronchiolitis and, hence, the failure of this therapy to impact on the clinical outcomes may be attributed to the lack of impact on the underlying process. Alternatively, it is possible that insufficient saline is delivered to the distal conducting airways due to airflow obstruction resulting from luminal secretions and mucosal oedema. This seems unlikely, as we used a modern nebuliser specifically designed for use in this age group which was well tolerated, and the frequency of therapy was greater than that used in some previous studies. While crying or distress may adversely affect drug delivery,29 it was reported that infants tolerated the treatment extremely well. This was probably related to their age, distress being more common in older infants and toddlers.
The results of this trial conflict with the conclusions of a Cochrane review, for which searches were last updated in April 2013–May 2013.20 The Cochrane team's meta-analysis of five trials (n=500) suggested HS-reduced LoS by more than 1 day (mean difference −1.15, 95% CI −1.49 to 0.82 days) with moderate levels of heterogeneity (I2=30). The summary effect size and associated heterogeneity were both driven by two studies, whose effect sizes made them outliers, each of which demonstrated CIs two-thirds the size of SABRE's from sample sizes of 93 and 112. Our own systematic review (PROSPERO CRD42014007569), which has different inclusion criteria, reckon there to be six eligible studies published since the Cochrane group updated their searches and a further three not found by the Cochrane review. Their synthesis will allow for a more reliable estimate of summary effect and heterogeneity as well as for a proper investigation of publication bias. None of these studies reported a reduction in the LoS. This includes the biggest study to date apart from SABRE,30 which also reported a null result (−0.02, 95% CI −0.25 to 0.21 days). A further study involving 160 hospitalised infants from Holland also reported no difference in duration of hospitalisation when comparing 6% HS, 3% HS and normal saline.31
One issue that may have contributed to the differences reported in the outcomes between this study and the Cochrane review might be the use of the term ‘acute bronchiolitis’ which can vary between countries, as noted in the introduction. The patients recruited to this study met the standard British definition with evidence of viral infection, airways obstruction and widespread crackles in the chest with or without wheeze. Studies included in the Cochrane review specify that they included infants with a viral infection and wheeze. Hence, it is possible that they may have included patients with similar but different underlying pathologies. However, studies have demonstrated the lack of precision in the use of the term ‘wheeze’,32 ,33 and in itself is unlikely to be a major issue particularly as the inflammation observed in those with acute bronchiolitis and ‘wheezy bronchitis’ is likely to be very similar. Of those studies included in the Cochrane review addressing the impact on LoS, three studies included subjects with a median age of 4.3 months or less suggesting that they were a very similar population to those included in this study. Two Chinese studies included patients with a median age of 5.8 months and in a Canadian study this was 8.9 months. The inclusion of subjects older than a year of age in these studies may have resulted in the inclusion of a minority of asthmatic subjects though the impact of this on the results is unclear. The patients in our study were well characterised, and the results can be confidently applied to the UK healthcare system.
In summary, this large, multicentre, open, randomised trial failed to demonstrate any impact on any of the clinically relevant outcomes when nebulised 3% HS administered 6-hourly was compared with good supportive care with minimal handlings in acute severe bronchiolitis. It did not appear to be responsible for a significant number of adverse events.
Acknowledgments
Trial Management Group: DH, EC, Colin O'Keeffe, Kirsty Sprange, CM, (University of Sheffield); JF (University of Leeds); MB, Simon Dixon, (University of Sheffield); MLE, (The University of Western Australia); KU, Heather Elphick, Sarah Shortland, Wendy Swann, (Sheffield Children's NHS Foundation Trust); JA, Kate Leech, Joanne Tomlinson, Marie Phipps, Ruth Jones, Eric Roe, Helen Parker, (University Hospital of North Staffordshire NHS Trust); Philip AJ Chetcuti, Louise Blackburn, Rebecca Mottram, Marjorie Allen, Nicola Balatoni, (Leeds Teaching Hospitals NHS Trust); Eduardo F Moya, Kelly Young, Ruth Skelton, Louise Akeroyd, Heather Rostron, (Bradford Teaching Hospitals NHS Foundation Trust); Lavleen Chadha, Mathew Kurian, Janet Queen, Sarah Shortland, (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Jonathan P Garside, Rachel Swingler, (Calderdale and Huddersfield NHS Foundation Trust); Colin Powell, Caroline Amphlett, Pauline Jones, (University Hospital of Wales); Ravinderjit S Lehal, Jennifer McKenna, Louise Willis, Rebecca Beckley, (Oxford University Hospitals NHS Trust); PSMN, Ian Sinha, Gail Wallace, Hannah Leyland, Timothy Henderson, Karen Phelan, (Alder Hey Children's NHS Foundation Trust); Peter I MacFarlane, Sarah Shortland, (Rotherham NHS Foundation Trust).
References
Footnotes
Trial Steering Committee: Jonathan Grigg, (Barts and The London, School of Medicine and Dentistry); Eirini Koutoumanou, (University College London); Jeremy Hull, (Oxford University Hospitals NHS Trust); Penny Broadley, (Sheffield Children's NHS Foundation Trust); MLE, KU, (Sheffield Children's NHS Foundation Trust); EC, Colin O'Keeffe, Kirsty Sprange, (University of Sheffield); Wendy Swann, (Sheffield Children's NHS Foundation Trust); Alexa Cross, (Health Technology Assessment); JF, (University of Leeds).
Data Monitoring and Ethics Committee: Tim Cole, (University College London); Neil Gibson, (Royal Hospital for Sick Children), Glasgow; Jayesh Bhatt, (Nottingham University Hospitals NHS Trust).
Co-applicants: MLE, (The University of Western Australia); CLC, Simon Dixon, (University of Sheffield); JF, (University of Leeds); Warren Lenney, (University Hospital of North Staffordshire NHS Trust); Philip AJ Chetcuti, (Leeds Teaching Hospitals NHS Trust); Eduardo F Moya, (Bradford Teaching Hospitals NHS Foundation Trust); Matthew Kurian, (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Jonathan P Garside, (Calderdale and Huddersfield NHS Foundation Trust); Peter I MacFarlane, (Rotherham NHS Foundation Trust); Heather Elphick, (Sheffield Children's NHS Foundation Trust); Colin Powell, (University Hospital of Wales).
Local Investigators: MarkLE, (The University of Western Australia); KU, Heather Elphick, (Sheffield Children's NHS Foundation Trust); JA, (University Hospital of North Staffordshire NHS Trust); Philip AJ Chetcuti, (Leeds Teaching Hospitals NHS Trust); Eduardo F Moya, (Bradford Teaching Hospitals NHS Foundation Trust); Lavleen Chadha, Matthew Kurian, (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Jonathan P Garside, (Calderdale and Huddersfield NHS Foundation Trust); Colin Powell, (University Hospital of Wales); Ravinderjit S Lehal, (Oxford University Hospitals NHS Trust); PSMN, (Alder Hey Children's NHS Foundation Trust); Peter I MacFarlane, (Rotherham NHS Foundation Trust).
Systematic Review team: CM, DH, HC, (University of Sheffield); MarkLE, (The University of Western Australia).
Pharmacy Staff: John Bane, Susan George and Jayne Clements, (Sheffield Children's NHS Foundation Trust); Catrin Barker, Carol Craig, Gill Swift, (Alder Hey Children's NHS Foundation Trust); Christine Brighouse-Johnson, (Calderdale and Huddersfield NHS Foundation Trust); Mark Fairweather, (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Susan Brammer, Paul Hollinshead and Carole Beardmore, (University Hospital of North Staffordshire NHS Trust); Kathryn Bethune, Annette Stone and Annie Rainey, (University Hospital of Wales); Sophia Khan, (Bradford Teaching Hospitals NHS Foundation Trust); Caroline Bedford, Paula Smalley, Helen Cunliffe and Sian Collins, (Leeds Teaching Hospitals NHS Trust); Laura Watson, (Oxford University Hospitals NHS Trust).
Study Monitoring: Joseph Clark, (University of Sheffield).
Data Management: Amanda Loban, (University of Sheffield).
Administrative and clerical support: Kylie Cross, (University of Sheffield).
Support on ethical and governance issues: Paul Dimitri and Gillian Gatenby, (Sheffield Children's Hospital NHS Foundation Trust).
Collaborators The SABRE Research Team The SABRE study team members: MLE, (The University of Western Australia); DH, (University of Sheffield); KU, (Sheffield Children's NHS Foundation Trust); JF, (University of Leeds); MB, Simon Dixon, CM, HC, (University of Sheffield); JA, Warren Lenney, (University Hospital of North Staffordshire NHS Trust); PSMN, (Alder Hey Children's NHS Foundation Trust); Heather Elphick, (Sheffield Children's NHS Foundation Trust); Philip AJ Chetcuti, (Leeds Teaching Hospitals NHS Trust); Eduardo F Moya, (Bradford Teaching Hospitals NHS Foundation Trust); Colin Powell, (University Hospital of Wales); Jonathan P Garside, (Calderdale and Huddersfield NHS Foundation Trust); Lavleen Chadha, Matthew Kurian, (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Ravinderjit S Lehal, (Oxford University Hospitals NHS Trust); Peter I MacFarlane, (Rotherham NHS Foundation Trust); CLC, EC, (University of Sheffield).
Contributors MarkLE, DH, KU, JF, MB, CLC, EC, CM, HC together produced the first draft of the publication. The following conceived of or designed the work: MLE, DH, JF, CLC. The following were involved in the acquisition of data for the work: MLE, KU. The following were involved in the analysis of data: JF, MB, CM, HC. MLE, DH, KU, JF, MB, CLC, CM, HC were involved in the interpretation of data for the work.
Funding This project was funded by the National Institute for Health Research Health Technology Assessment (HTA) Programme (project number 09/91/22).
Disclaimer The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the HTA Programme, NIHR, NHS or the Department of Health.
Competing interests PSMN has the following conflicts of interest: Paul Alios Biopharma—Personal fees—Consultancy payments for assisting in setting up study on new antiviral for RSV disease in the UK 2013–2015. Janassen Pharaceuticals—Personal fees—Honorarium received for advisory board attendance to discuss new RSV antivirals and vaccines (February 2014). We declare that all remaining authors have no conflicts of interest.
Ethics approval NRES Committee Yorkshire & Humber – South Yorkshire.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Requests for further data not available in this publication can be directed at Sheffield Clinical Trials Research Unit. Email: ctru@sheffield.ac.uk Tel: 0114 222 0870.