Article Text

Abstract
Rationale Newborn screening (NBS) for cystic fibrosis (CF) allows early intervention. Design of randomised controlled trials (RCT) is currently impeded by uncertainty regarding evolution of lung function, an important trial end point in such infants.
Objective To assess changes in pulmonary function during the first year of life in CF NBS infants.
Methods Observational longitudinal study. CF NBS infants and healthy controls were recruited between 2009 and 2011. Lung Clearance Index (LCI), plethysmographic lung volume (plethysmographic functional residual capacity (FRCpleth)) and forced expired volume (FEV0.5) were measured at 3 months and 1 year of age.
Main results Paired measurements were obtained from 72 CF infants and 44 controls. At 3 months, CF infants had significantly worse lung function for all tests. FEV0.5 improved significantly (0.59 (95% CI 0.18 to 0.99) z-scores; p<0.01) in CF infants between 3 months and 1 year, and by 1 year, FEV0.5 was only 0.52 (0.89 to 0.15) z-scores less than in controls. LCI and FRCpleth remained stable throughout the first year of life, being on average 0.8 z-scores higher in infants with CF. Pulmonary function at 1 year was predicted by that at 3 months. Among the 45 CF infants with entirely normal LCI and FEV0.5 at 3 months, 80% remained so at 1 year, while 74% of those with early abnormalities remained abnormal at 1 year.
Conclusions This is the first study reporting improvements in FEV0.5 over time in stable NBS CF infants treated with standard therapy. Milder changes in lung function occurred by 1 year than previously reported. Lung function at 3 months predicts a high-risk group, who should be considered for intensification of treatment and enrolment into RCTs.
- Cystic Fibrosis
- Lung Physiology
- Paediatric Lung Disaese
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
Newborn screened cystic fibrosis (CF) infants have abnormal lung function by 3 months of age; how does this change during the first year of life?
What is the bottom line?
-
Lung function remained stable or improved in newborn screened CF infants during the first year of life; deficits at 1 year were considerably smaller than previously documented in either screened or clinically diagnosed infants.
Why read on?
-
This study, the largest of its kind and the only one with contemporaneous healthy controls, describes early lung development in newborn screened infants with CF; these data will inform the design of interventional trials in these children.
Introduction
The major cause of morbidity and mortality in cystic fibrosis (CF) is pulmonary disease. Until recently, treatment has been targeted at the downstream consequences of CFTR dysfunction, such as bronchial infection, inflammation and mucus retention. A recent paradigm shift has, however, led to development of genotype class-specific therapies, such as PTC124 to over-ride premature stop codons1 ,2 and VX-770 for the class 3 mutation G551D.3 ,4 It seems likely that these novel therapies will be most effective in early stage disease, before irreversible airway damage has developed. It is therefore essential to understand the evolution of lung function in newborn screened (NBS) CF infants given standard treatment, in order to determine optimal trial endpoints and adequately power intervention studies.
CF infants diagnosed clinically have airflow obstruction at diagnosis, even in the absence of respiratory symptoms, signs or history of infection,5 with no improvement in pulmonary function over the ensuing years despite specialist treatment.6–8 CF NBS has been introduced in the hope that earlier diagnosis will lead to improved outcomes. Previous longitudinal studies investigating lung function in NBS CF infants reported progressive decline in the early years, despite specialist treatment.9 ,10
Following recent universal introduction of screening throughout the UK, we recruited a cohort of NBS infants with CF and healthy controls between 2009 and 2011. Disappointingly, only 56% of those with CF had normal pulmonary function tests (PFT) when assessed at 3 months.11 The current manuscript describes follow-up PFTs at 1 year for this cohort. Our primary hypothesis was that lung function would deteriorate further between 3 months and 1 year of age. We also aimed to investigate the determinants of lung function at 1 year, and to collect data to assess feasibility of recruiting NBS CF infants to invasive studies12 and inform future power calculations.
Methods
NBS CF infants born between January 2009 and July 2011 who were referred to the six specialist CF centres in the London CF Collaboration (LCFC) were eligible for recruitment.11 Healthy controls were recruited contemporaneously from Homerton University Hospital, East London. Infants were ineligible if born <36 weeks gestation or had coexisting congenital abnormalities (see online supplementary data). The study was approved by the North Thames Multi-Centre Research Ethics Committee (#09/HO71/314). Informed written parental consent was obtained.
Participating centres prospectively completed Case Record Forms (CRF) at diagnosis and each subsequent clinic visit (see online supplementary data). CF infants were started on multivitamins and vitamin E, pancreatic enzyme replacement therapy where appropriate and, in accord with UK CF Trust guidelines, prophylactic flucloxacillin, according to a standardised treatment protocol (see online supplementary data).
Infant PFTs
All infants were tested at Great Ormond Street Hospital/UCL Institute of Child Health at around 3 months and 1 year postnatal age. Infants were free of respiratory illness for at least 3 weeks before PFTs. Infants were weighed and examined prior to administering chloral hydrate orally or rectally (60–100 mg/kg). Weight and crown-heel length were expressed as z-scores to adjust for age and sex.13 Heart rate and SpO2 were monitored continuously throughout testing. Infant urine or maternal saliva samples were collected for cotinine assay to validate maternal report of smoking. PFTs were undertaken according to international guidelines.14 ,15 Lung Clearance Index (LCI), a measure of ventilation inhomogeneity was measured by multiple breath washout (MBW), using mass spectrometry and customised software.16 Plethysmographic Functional Residual Capacity (FRCpleth) and forced expired volumes (FEV0.5) and flows (FEF75) from an inflation pressure of 30 cm H2O using the raised volume technique were measured using the Jaeger BabyBody device (CareFusion, San Diego, USA; V.4.65).15 PFT results were electronically exported to a research database (Re-Base software, Re-Base, UK), which contained all relevant demographic and clinical details. PFT results were expressed as z-scores to adjust for body size, sex and age, using reference equations derived from healthy infants studied with identical equipment and protocols.11 ,17–19 Abnormal PFTs were defined as results outside the 95% limits of normal: that is, >1.96 z-scores (>97.5th centile) for LCI and FRCpleth or <−1.96 z-scores (<2.5th centile) for FEV0.5. Results were reported to the physicians responsible for the clinical care of each child, and subsequently discussed with parents.
Statistical analysis
Data were inspected for distribution and calculation of descriptive statistics (PASW Statistics V.18, Chicago, Illinois, USA). Significance was taken as p<0.05. Lung function results at 3 months, at 1 year and changes between 3 months and 1 year were compared between groups using Student t-test. Multivariable linear regressions were used to investigate how lung function variables at 1 year, and change in lung function between 3 months and 1 year, varied according to potential determinants (background characteristics, clinical symptoms, antibiotic treatment and microbiological results, see online supplementary data for details). Model estimates and differences between groups are presented with 95% CIs. Multiple imputations were used to impute values for any failed PFTs at 3 months (see online supplementary data). Taking into account three primary outcomes (LCI, FRCpleth and FEV0.5), a sample size of 72 infants with CF and 44 controls at 1 year (equivalent to 53/group if equal groups) allows detection of differences between groups equivalent to 0.66 z-scores at the 5% significance level with 84% power.20–22
Results
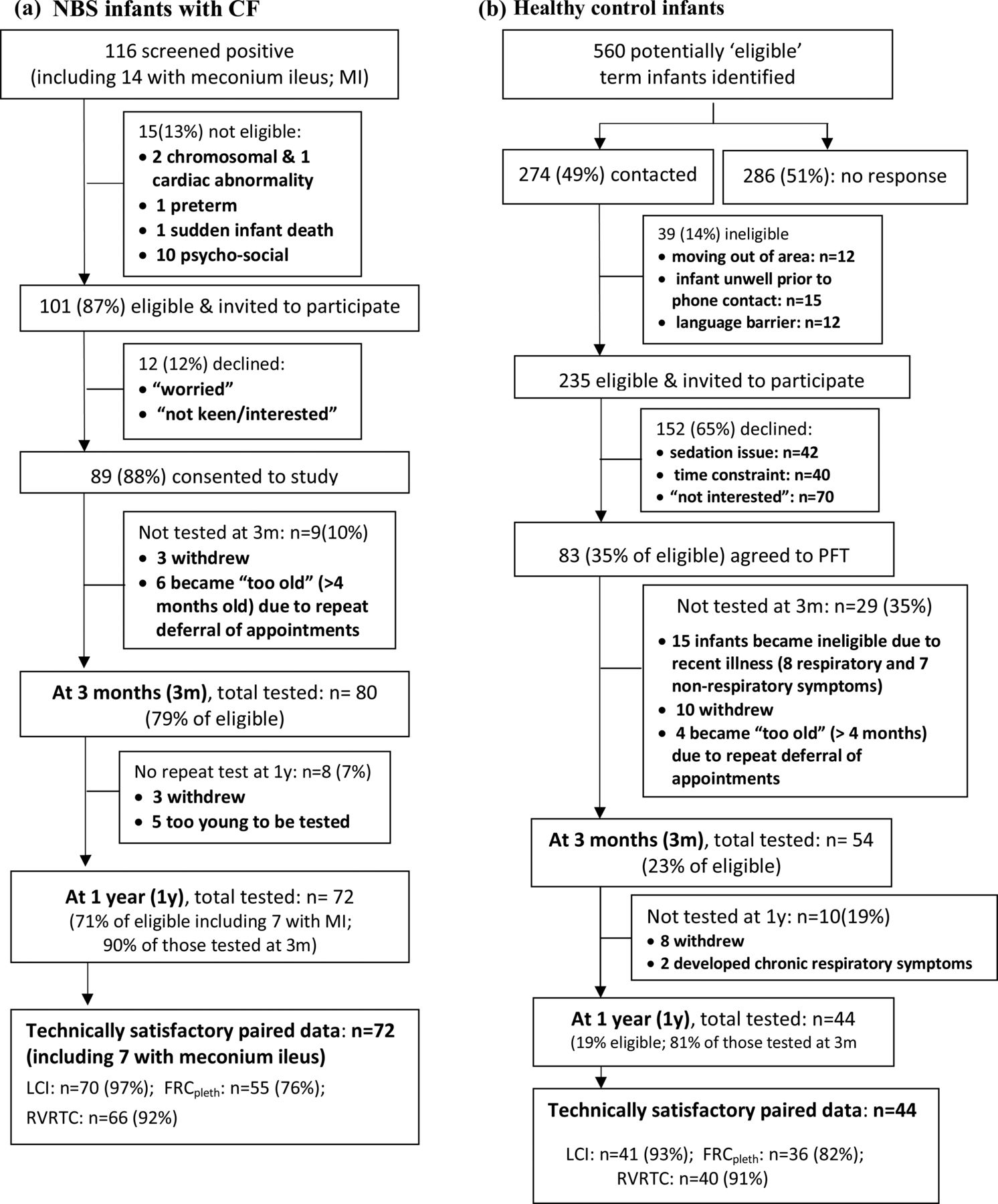
The screening, recruitment and follow-up of subjects are shown in figure 1. Paired measurements at 3 months and 1 year were obtained from 72 of 101 CF NBS infants, (90% of those tested at ∼3 months of age). Inspection of CRFs and regular communication with consultants revealed excellent adherence to treatment protocols. Details of additional treatment are provided in the online supplementary data. Paired measurements were obtained from 44 contemporaneous controls (81% of those tested at 3 months). CF infants were born slightly earlier with lower birth weight than controls, but background characteristics were otherwise similar (table 1). There was no difference between groups regarding change in weight between birth and first PFTs at ∼3 months (mean difference CF-controls: −0.14 (95% CI −0.56 to 0.29) weight z-scores).
Characteristics of CF and healthy controls infants with paired lung function at 3 months and 1 year

Success rates for recruitment and achievement of technically acceptable infant pulmonary function data. NBS, newborn screened; PFTs, pulmonary function tests; LCI, lung clearance index; FRC, functional residual capacity; MBW, multiple breath inert gas washout; pleth, plethysmographic technique; RVRTC,Raised Volume Rapid Thoraco-abdominal Compression, from which forced expired flows and volumes were derived. See online supplementary table E1 for details of PFT success on each test occasion.
For CF NBS infants, the median (IQR) age at diagnosis was 3.6 (3.0–4.4) weeks with 7 (10%) infants presenting with meconium ileus. Since inclusion of these infants did not affect the results (data not shown), they were included in the analysis. At 1 year PFTs, cough-swab cultures had been positive on at least one occasion for Pseudomonas aeruginosa in 25 (35%), and for other significant bacteria in 17 (24%).
Anthropometry and lung function results
Comparison between infants with CF and healthy controls
Success in obtaining technically satisfactory PFTs were similar between groups, but varied by age and outcome, being lowest for FRCpleth at 3 months (76%) and highest for LCI (≥93% on both occasions; figure 1 and see online supplementary table E1). At 3 months, CF infants had significantly lower weight, height and body mass index (BMI); higher LCI and FRCpleth; and lower FEV0.5, forced vital capacity (FVC) and FEF75 compared with controls (table 2). Significant increases in z-scores for somatic growth were observed in both groups between 3 and 12 months, but changes were significantly greater in CF infants, such that there were no between-group differences by 1 year. All PFT results remained stable in healthy infants during the first year of life, as did LCI, FRCpleth and FVC in those with CF. However, FEV0.5 and FEF75 z-scores improved between test occasions in CF infants (table 2, see online supplementary figure E1): differences in FEF75 no longer being significant by 1 year when compared with controls. Although not one of the selected primary outcomes, there was a significant increase in gas trapping (as reflected by ΔFRC, ie, the within-subject difference in FRC assessed using plethysmography and MBW) in CF infants during the first year of life (table 2).
Comparison of anthropometry and pulmonary function at ∼3 months and 1 year in CF NBS infants and healthy controls (HC)
Determinants of lung function at 1 year
On linear univariable analysis, LCI, FRCpleth and FEV0.5 z-scores at 1 year were significantly associated with CF status and 3 months PFT (see online supplementary table E3). Using multivariable linear regression, significant determinants of 1 year LCI z-score were: CF status (regression coefficient (95% CI) 0.48 (0.04 to 0.93)), 3 month LCI (0.24 (0.07 to 0.41) per unit z-score), history of clinician-diagnosed wheeze (0.59 (0.05 to 1.12)) and change in weight z-score between birth and first PFT (−0.18 (−0.35 to −0.01) per unit z-score). For 1 year FRCpleth z-score, determinants were: 3 months FRCpleth (0.43 (0.27 to 0.59) per unit z-score), history of PsA infection (0.71 (0.24 to 1.17)) and change in weight z-score between 3 and 12 months (−0.20 (−0.41 to 0.0) per unit z-score change), whereas 1 year FEV0.5 z-score was only significantly associated with 3 months FEV0.5 on multivariable analysis (−0.18 (−0.35 to −0.01) per unit z-score).
Relationship between PFT results at 3 months and 1 year
At 3 months of age, LCI, FRCpleth and FEV0.5 were abnormal in 17% (12/71), 16% (9/57) and 26% (18/68) of CF infants, respectively. By 1 year, the percentage with abnormal FEV0.5 had decreased to 9% (6/69) (mean difference (95% CI) −18% (−30% to −5%)) whereas those with abnormal LCI (18% (13/71)) and FRCpleth (16%(11/70)) remained virtually unchanged. Significant correlations were found between PFTs at 3 months and 1 year (figure 2 and see online supplementary table E2). Of the 52 infants in whom all three PFTs were technically successful on both occasions, abnormalities were observed in 33% (17/52) at first test, 10 (59%) of whom remained abnormal at follow-up. Of the 35 (67%) with entirely normal results at 3 months 25 (71%) remained so at 1-year.
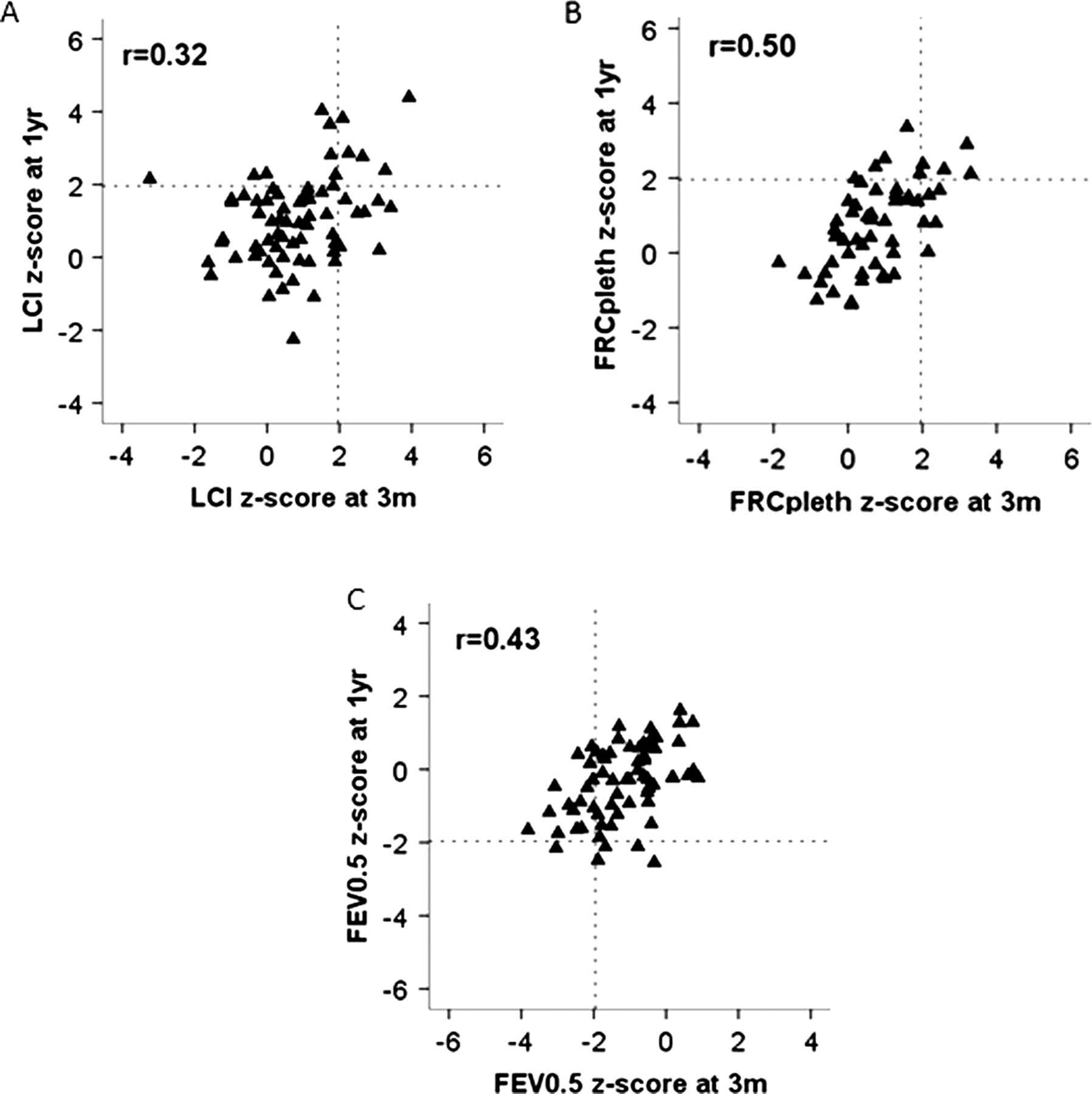
Relationship between pulmonary function at 3 months and 1 year in newborn screened CF infants. The 95% limits of ‘normal range’ (97.5th centile for Lung Clearance Index (LCI) and functional residual capacity (FRC) and 2.5th centile for FEV0.5) are represented by vertical dashed lines at 3 months (3m) and horizontal lines at 1 year (1yr). Those with normal pulmonary function tests on both occasions fall within the lower left quadrant for LCI and FRC, and upper right quadrant for FEV0.5. Infants with abnormal LCI at 3 months but normal LCI at a year, lie within the lower right quadrant (A), while those with abnormal FEV0.5 at 3 months which has normalised by 1 year are within the left upper quadrant of (C).
Identification of a high risk group of NBS CF infants
Based on results from LCI and FEV0.5 which were the most feasible outcomes at 3 months (see online supplementary table E1), we attempted to delineate a subgroup of infants who would be at high risk of having abnormal lung function at 1 year, and who thus might be suitable candidates for an intervention study. Among the 64 CF infants in whom acceptable LCI and FEV0.5 results were obtained on both occasions, abnormalities (elevated LCI or diminished FEV0.5) were identified in 19 (30%) at ∼3 months, of whom 14 (74%) remained abnormal at 1 year. Among CF infants with entirely normal PFTs at 3 months from these two tests (n=45), 36 (80%) remained so at 1 year (see online supplementary data for details). There were no significant differences at 1 year in FEV0.5 (−0.36 (−0.9 to 0.17) z-scores) or LCI (0.46 (−0.13 to 1.05) z-scores) between CF infants with normal 3 months PFTs and healthy controls. By contrast, when compared with controls at 1 year, LCI was 1.33 (0.6 to 2.1) z-scores higher and FEV0.5 −0.8 (−1.5 to −0.1) z-scores lower in those with abnormal PFTs by 3 months (see online supplementary table E3).
Discussion
Contrary to our hypotheses, forced expired flows and volumes improved by 1 year of age, with stability of other PFTs in NBS CF infants. This is the first time such improvement has been reported in an observational longitudinal study of NBS CF infants. The number of CF infants with abnormal LCI and FRCpleth at 1 year was similar to that at 3 months, while there was a significant reduction in those with abnormal FEV0.5 during this period. Impaired lung function at 1 year was predicted by lung function at 3 months and associated with clinician-diagnosed wheeze (LCI), poor weight gain (LCI and FRCpleth) and prior P aeruginosa (FRCpleth).
Strengths and limitations
The major strengths of this study are that longitudinal assessments of lung function were undertaken in a large cohort of NBS CF infants within a single location, results being directly compared with healthy controls. Attrition was minimal,12 with no bias between those who did and did not complete the study. Selection of various PFTs enabled different aspects of pathophysiology to be assessed.11 Appropriate reference equations for infant PFTs,18 ,19 including LCI, which has now been shown to be dependent on body size during early life,17 facilitated accurate interpretation of results. Limitations are that, in an observational study such as this, we can only demonstrate association not causation of potential determinants of 1 year lung function. Computed tomography (CT) and broncho-alveolar lavage were performed at 1 year in CF infants, but not at 3 months, and are therefore not reported in this paper, which focusses on longitudinal changes. Furthermore, structural changes on CT at 1 year were very mild and poorly reproducible.25
Interpretation of PFTs
As reported previously,16 since the infant PFTs were selected to reflect a wide a range of lung pathology, the relatively poor correlations between the different primary outcomes on any one test occasion (see online supplementary table E2) was not surprising. While spirometry is known to be less sensitive than LCI for detection of mild lung disease in preschool children with CF,8 during infancy FEV0.5 has been shown to be a sensitive outcome in clinically diagnosed CF infants.16 While this was also observed in this study of NBS infants at 3 months of age,11 by 1 year far fewer NBS infants were identified by the raised volume technique than either plethysmography or LCI. This may reflect the mild nature of lung disease at 1 year in our NBS cohort when compared with those diagnosed clinically and the decreasing sensitivity of forced expiratory manoeuvres to mild lung disease as airway and chest-wall compliance decrease with increasing maturity.26 By contrast with the lack of correlation between FEV0.5 and other lung function outcomes on either test occasion, there were significant associations between LCI, FRCpleth and ΔFRC, all of which are thought to be sensitive measures of peripheral airway disease throughout childhood (see online supplementary table E2). Whatever the interpretation of these changes, as discussed below, they are in sharp contrast with those previously reported in CF infants. Consequently, when selecting outcome measures for intervention trials in NBS CF infants,27 reliance should not be placed solely on the raised volume technique, since measures of LCI appear essential if mild abnormalities are to be detected. While hyperinflation and gas trapping also proved to be sensitive outcomes at 1 year, routine inclusion of these outcomes shortly after birth may be limited by equipment costs and increased failure rate of FRCpleth in young infants. With the exception of a significantly lower FEV0.5 (mean (95% CI): −0.70 (−1.29 to −0.10) z-scores) in those who received additional antibiotics for symptoms or positive cough swab, there was no significant association between PFT outcomes and the infants’ genotype, clinical status or any acute interventions prior to PFTs at 3 months.11
Comparison with the literature
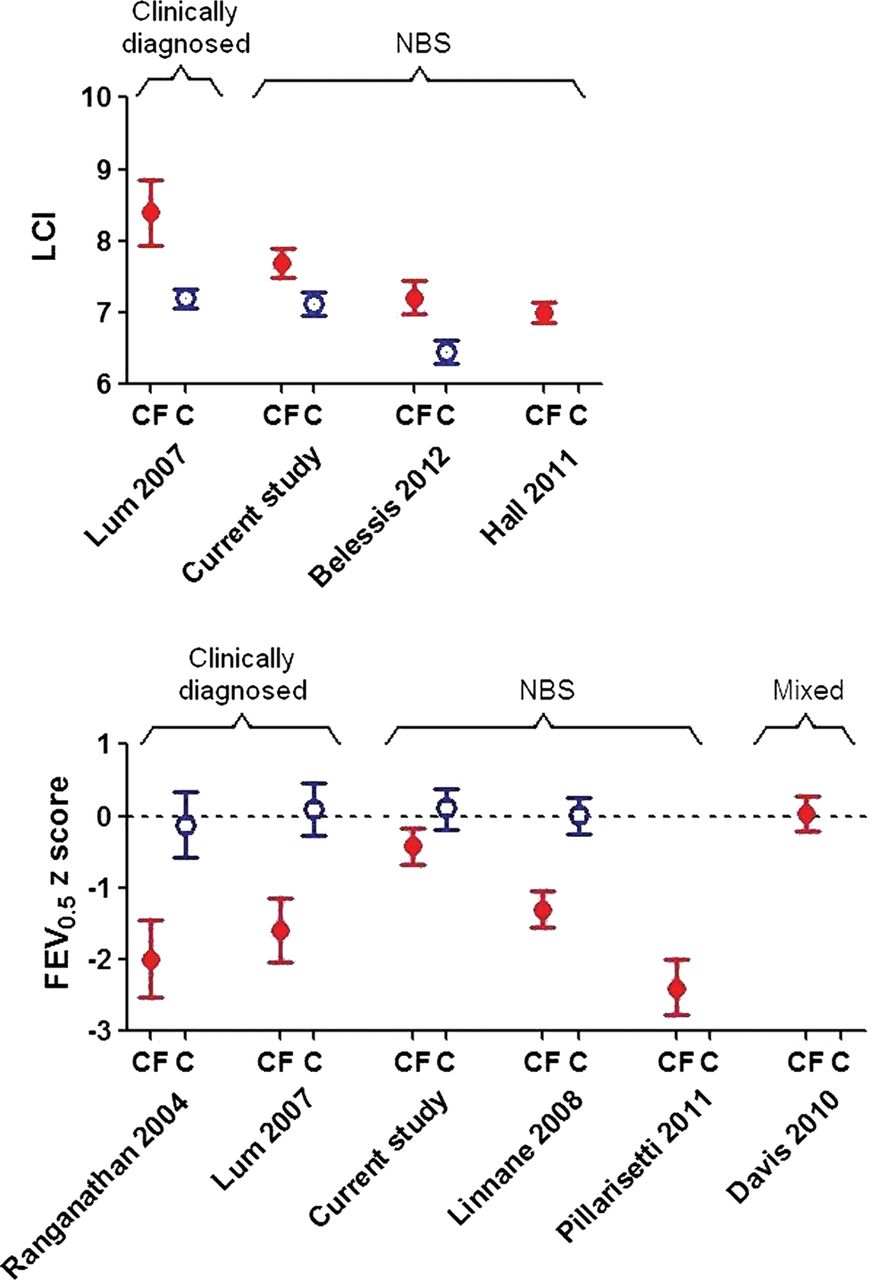
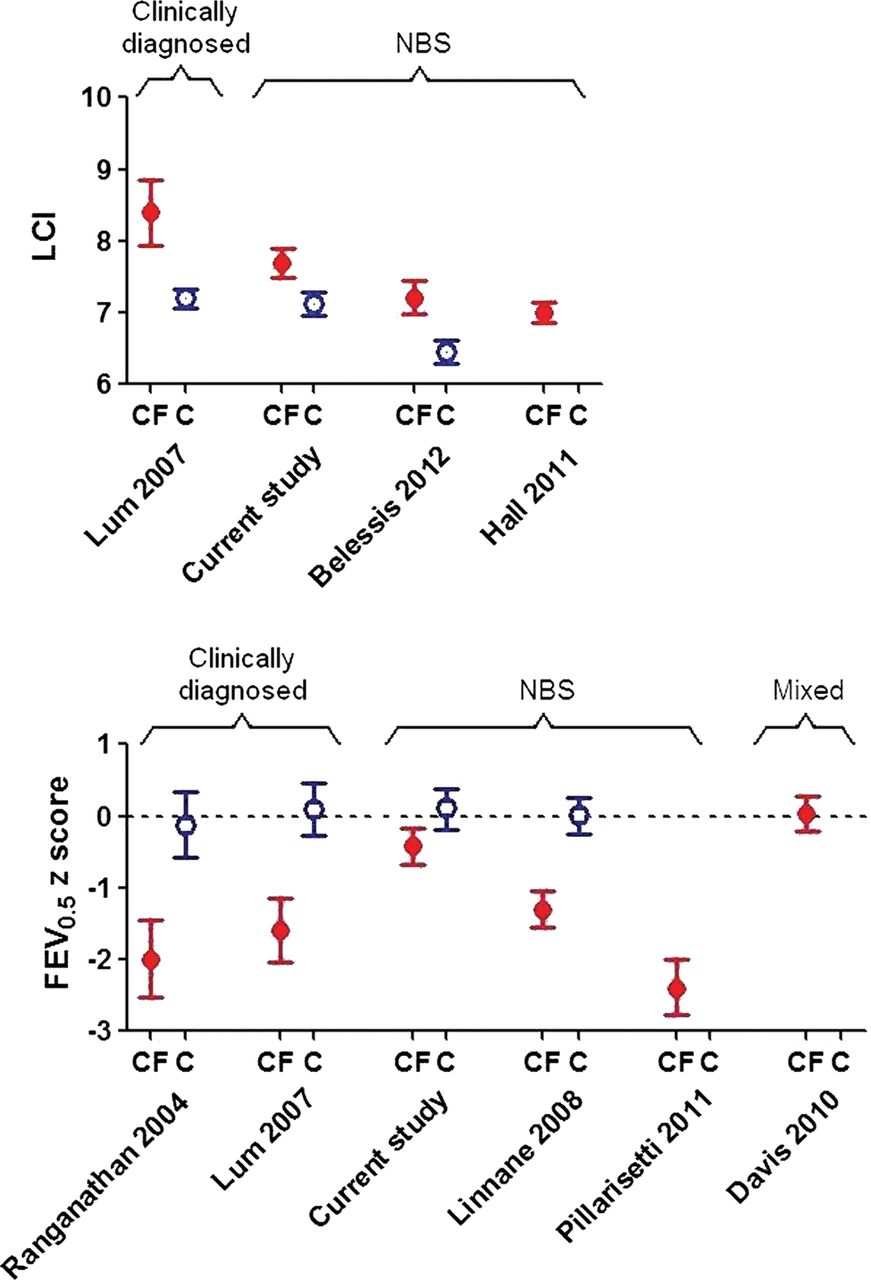
Results regarding evolution of early lung disease in those diagnosed by NBS have been conflicting (figure 3). The Australian Respiratory Early Surveillance team for CF (AREST-CF) have reported normal and reduced PFTs in such infants within the first 6 months of life,9 with further rapid deterioration over the first year of life (mean FEV0.5 being −2.4 z-scores by ∼1 year of age).10
{kind=link}
{kind=link}
{kind=link}
Comparison of current lung function results in infants with cystic fibrosis (CF) and healthy controls (C) at ∼1 year of age, with previously published results. Data expressed as mean (95% CI). To allow direct comparison with previously published studies, Lung Clearance Index is presented in absolute units, whereas FEV0.5 is expressed as z-scores, based on different reference equations according to each author. The dashed horizontal line at 0 z-scores equates to 100% predicted based on a healthy population. Control data were not available in all studies. NBS, newborn screening.
In the current study, lung function was abnormal by 3 months,11 but stabilised or improved thereafter. As can be seen from figure 3, 1 year-lung function in the LCFC NBS cohort was significantly better than that in previous clinically diagnosed LCFC cohorts6 ,16 or in the AREST-CF NBS cohort at similar age.9 ,10 The reasons for the discrepancies between our results and those for AREST-CF are unclear. While the standardised protocol adhered to by the LCFC differs in some respects from that used by most centres in the USA, Australia and Europe (eg, use of flucloxacillin prophylaxis), the results should be a benchmark for other centres, and could serve as the basis for quality improvement.28 Median age at first test in this study is younger than that in AREST-CF, which may reflect earlier diagnosis and implementation of treatment within the narrow geographical area of southeast England that we recruited from, thereby halting progression of any early lung disease. It is possible that infants recruited to AREST-CF were sicker, or deteriorated faster due to differences in modifier genes, environment or adherence to treatment, when compared with those in London. Most importantly, by contrast with the current study, AREST-CF data were not compared with contemporaneous controls, historical controls being used initially,9 with subsequent results (obtained using higher inflation pressures10) being interpreted using reference data based on different equipment, which can bias interpretation.19 ,29
Improvements in lung function following treatment for acute exacerbations in infants with CF have been demonstrated, 30 but ours is the first study to document improvements in FEV0.5 in infants treated with standard therapy, studied during periods of clinical stability. A recent exploratory study reported greater increases in FEV0.5 over a 48-week period in 22 infants and young children treated with hypertonic saline compared with 23 randomised to isotonic saline (mean (95% CI) difference:38 (1 to 76) mL).31 However, from the data presented, it is impossible to ascertain whether this reflected stability, improvement or simply less deterioration over time with active treatment, once effects of lung and somatic growth had been accounted for.
Clinical implications
These results have implications for clinical practice and research. Although PFTs represent only one of the potential outcomes that can be used during early life,27 with additional information gleaned from inflammatory markers and computerised tomography,32 they represent the mainstay of clinical management and a major outcome in randomised controlled trials (RCTs) in children and adults. Since lung function tracks from late infancy into later life, accurate identification of early abnormalities is imperative. Furthermore, given the increasing number of centres undertaking ‘clinical’ infant PFTs,33 the current study may facilitate more meaningful interpretation of results by providing vital evidence regarding the natural changes that can occur over time in healthy infants and those with lung disease, in the absence of any specific interventions.
We have shown that lung function and somatic growth during the first year of life are significantly better in infants diagnosed by NBS in the UK than in their counterparts who were clinically diagnosed a decade earlier6 ,16 (figure 3). It is, however, of concern that despite early diagnosis and prompt treatment, LCI remains abnormal at 1 year (figure 3), albeit to a mild degree.16 Further follow-up is required to establish the extent to which these changes predict later outcome. Nevertheless, in this study, normal lung function was sustained in at least 50% NBS CF infants to 1 year of age. The significant improvement in FEV0.5 and stability of sensitive measures of distal airway function during early life when on ‘standard therapy’, and the relatively small deficits in lung function in CF NBS infants at 1 year also have important implications for design of future randomised intervention trials, which are essential to better define better standards of care in this age group. Despite considerable within-subject variability, the main predictor of lung function at 1 year was that at 3 months, allowing us to identify a ‘high-risk’ group who could potentially be targeted for future intervention trials.
Using data from this study, results from ∼85 infants/arm would be required to detect relatively small differences in lung function (ie, equivalent to 0.5 z-scores) that might occur in response to an intervention if unselected NBS CF were recruited to such a trial. By contrast, were recruitment to such a RCT limited to a ‘high-risk group’ (ie, abnormal PFTs by 3 months, see online supplementary tables E3 and E4), a larger treatment effect would be expected, with only 22 infants/arm being required to detect a difference of 1 z-score (equivalent to ∼9% for LCI), with 90% power. Such an approach could optimise recruitment since parents of infants with early PFT abnormalities would be more likely to consent, and also this approach would minimise exposure of children with potentially little to gain from therapy from unnecessary side effects.
In summary, we have shown that some measures of pulmonary function improve in the year following CF NBS diagnosis, and none deteriorate. Performing randomised intervention studies in an unselected cohort of infants using PFTs as an end point will, therefore, require large sample sizes due to the generally mild changes in lung function observed. Nonetheless, it is possible to identify CF infants with abnormal lung function by 3 months, who represent a high-risk group for persistent abnormalities at 1 year, and who may benefit from additional treatment during the vital first few years of life.
Acknowledgments
We thank the infants and parents who participated in this study, and gratefully acknowledge contributions by all members of the London NBS CF Collaboration (Ah-Fong Hoo, Ammani Prasad, Andrew Bush, Angie Wade, Anu Shankar, Catherine Owen, Caroline Pao, Colin Wallis, Deeba Ahmed, Gary Ruiz, Hilary Wyatt, Ian Balfour-Lynn, Jane Chudleigh, Jane Davies, Janet Stocks (Director), John Price, Lena Thia, Lucy Brennan, Mark Rosenthal, Paul Aurora, Ranjan Suri, Richard Chavasse, Siobhan Carr, Sooky Lum and The Thanh Diem Nguyen) and Per Gustafsson for on-going advice and support with respect to MBW by Mass spectrometry.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
TT-DN and LPT contributed equally.
-
Contributors JS and AB were responsible for the conception and design of the study; JS is responsible for supervision of the study and together with JC, for research governance issues including ethics committee approval; A-FH provided technical training, supervision and audit of data collection and analyses; A-FH and JC set up the recruitment process. Infants with CF were recruited by the paediatric respiratory consultants participating in the LCFC, including AB and PA. TT-DN, LPT, A-FH, JC and SL recruited the healthy infants, undertook all lung function measurements and, together with JS, calculated and interpreted lung function results; TT-DN, LPT and AW performed statistical analyses; TT-DN, LPT, AB, PA and JS drafted the manuscript; all remaining authors revised and approved the manuscript for intellectual content before submission.
-
Funding This study is supported by grants from the Cystic Fibrosis Trust, UK; Special Trustees: Great Ormond Street Hospital for Children, London, UK; Smiths Medical Ltd, UK; Comprehensive Local Research Network, UK. It was also supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval North Thames Multi-Centre Research Ethics Committee (#09/HO71/314).
-
Provenance and peer review Not commissioned; externally peer reviewed.