Article Text

Abstract
Background The mechanism underlying respiratory virus-induced cough hypersensitivity is unknown. Upregulation of airway neuronal receptors responsible for sensing physical and chemical stimuli is one possibility, and the transient receptor potential (TRP) channel family are potential candidates. We have used an in vitro model of sensory neurons and human rhinovirus (HRV-16) to study the effect of virus infection on TRP expression.
Methods IMR-32 neuroblastoma cells were differentiated in culture to express three TRP channels: TRPV1, TRPA1 and TRPM8. Flow cytometry and qRT-PCR were used to measure TRP channel protein and mRNA levels following inoculation with live virus, inactivated virus, virus-induced soluble factors or pelleted virus particles. Multiplex bioassay was used to determine nerve growth factor (NGF), interleukin (IL)-1β, IL-6 and IL-8 levels in response to infection.
Results Early upregulation of TRPA1 and TRPV1 expression occurred 2–4 h post infection. This was independent of replicating virus as virus-induced soluble factors alone were sufficient to increase channel expression 50-fold and 15-fold, respectively. NGF, IL-6 and IL-8 levels, increased in infected cell supernatants, represent possible candidates. In contrast, TRPM8 expression was maximal at 48 h (9.6-fold) and required virus replication rather than soluble factors.
Conclusions We show for the first time that rhinovirus can infect neuronal cells. Furthermore, infection causes upregulation of TRP channels by channel-specific mechanisms. The increase in TRPA1 and TRPV1 levels can be mediated by soluble factors induced by infection whereas TRPM8 requires replicating virus. TRP channels may be novel therapeutic targets for controlling virus-induced cough.
- Asthma
- Asthma Mechanisms
- Asthma Pharmacology
- Cough/Mechanisms/Pharmacology
- Cytokine Biology
- Respiratory Infection
- Viral infection
- Airway Epithelium
Statistics from Altmetric.com
- Asthma
- Asthma Mechanisms
- Asthma Pharmacology
- Cough/Mechanisms/Pharmacology
- Cytokine Biology
- Respiratory Infection
- Viral infection
- Airway Epithelium
Key messages
What is the key question?
-
What are the mechanisms responsible for respiratory virus-induced cough?
What is the bottom line?
-
Rhinovirus infection upregulates the expression of TRP channels, a group of neuronal receptors responsible for sensing physical and chemical stimuli. An increase in neuronal TRPA1 and TRPV1 levels can be mediated by soluble factors produced in response to rhinovirus infection whereas TRPM8 requires replicating virus.
Why read on?
-
This study provides new information on how respiratory viruses may modulate the airway sensory response and provides evidence for new therapeutic targets that may help control post-viral cough.
Introduction
Human rhinovirus (HRV) accounts for the majority of viral infections associated with the common cold1 and is an important cause of asthma exacerbations.2–4 During viral exacerbation the airway reflexes responsible for cough and bronchospasm become hypersensitive5 ,6 and exposure to chemical (eg, cigarette smoke, acidic air pollutants) and thermal (cold air) irritants can provoke bouts of troublesome cough and wheeze.
Although much is known about the acute inflammatory response to HRV infection,7–9 the precise mechanism for virus-induced airway reflex hypersensitivity has not yet been established. Virus-induced upregulation of receptors responsible for sensing chemical and physical stimuli is one possibility. The novel transient receptor potential (TRP) cation channel family, which is expressed in many cell types including airway sensory nerves,10–12 may be relevant as they are able to respond to irritant and thermal stimuli which provoke cough clinically. Several TRP channels, in particular TRP channel subfamily V member 1 (TRPV1), TRP channel melastatin member 8 (TRPM8) and TRP channel subfamily A member 1 (TRPA1), which are directly activated by chemical, thermal and mechanical stimuli, represent potential candidate receptors associated with respiratory virus-induced airway hypersensitivity. Both TRPA1 and TRPV1, expressed on vagal sensory nerves, are known to regulate the tussive response to airway irritants in both animal and human experiments.13 ,14 TRPA1, for example, has recently been considered a potentially important integrator in the neuroinflammatory responses in allergic asthma.10 ,11 Increased numbers of TRPV1-containing nerves have been reported in the airways of patients with idiopathic cough,15 many of whom recall a viral infection preceding the onset of their cough.16 A number of TRP receptor antagonists are already in clinical development,17 and insights into the role of these receptors in virus-induced cough may identify new therapeutic options.
Experimental infection of healthy human subjects with HRV-16 causes an infiltration of T cells into the submucosa in the intrapulmonary airways, which suggests a response to infection.18 ,19 Although this remains unproven, it is very possible that airway nerves may be a cell type susceptible to HRV infection as it has recently been shown that human airway parasympathetic nerves express the virus receptor intercellular adhesion molecule 1 (ICAM-1).20 Demonstration of HRV infection in cells modelling peripheral neurons would strongly indicate the potential for infection of airway nerves. The human neuroblastoma cell line IMR-32 can be differentiated (dIMR-32) to acquire neuronal morphology and physiology, making them highly characteristic of peripheral nerve cells. The most notable changes include outgrowth of long axon-like processes, selective expression of voltage gated calcium channels (almost exclusively N-type channels) and synthesis of neurotransmitters such as dopamine, norepinephrine and serotonin. They also show functional expression of TRP channels.21 In the present study we investigated whether HRV infection of the dIMR-32 cells in vitro upregulates the expression of neuronal TRP channels and, if so, whether this is mediated directed by replicating virus or indirectly by virus-induced soluble factors.
Materials and methods
Virus and cells
HRV-16 was purchased from the National Collection of Pathogenic Viruses (UK) and was grown and titred in a standard human lung fibroblast cell line (Wi-38 cells)22 at 33°C, as previously described.23 Wi-38 cells and the IMR-32 human neuroblastoma cell line were both purchased from the European collection of cell cultures (detailed in online supplement). Differentiated IMR-32 cells (dIMR-32) were derived according to the published protocol17 using 5 µM 5-bromo-2′-deoxyuridine (Sigma, UK). TRPA1 and TRPV1 transfected HEK cell lines (gifts from Dr Laura Sadofsky, Hull University) were grown in Dulbecco's modified Eagle's medium as described for HEK-TRPA1 cells,13 and the TRPM8 transfected HEK cell line (a gift from Professor Zholos, Queen's University Belfast) was grown as previously described.24
Infection and treatment of dIMR-32 cells
Cultures of dIMR-32 cells were infected with HRV at selected multiplicities of infection (MOI) to determine the optimum level of TRP expression. The cultures were mock infected with medium alone or treated with virus-free supernatant from infected Wi-38 cells. They were also treated with virus pellet (obtained by ultracentrifugation) and ultraviolet (UV)-inactivated virus preparations. These preparations were used to determine whether soluble factors present in the supernatant and/or direct interaction of virus particles with the neuronal cells are responsible for the modulation of TRP channels.
Immunofluorescence and flow cytometry
Antobodies used for immunofluorescence (IF) and/or flow cytometry were: TRP receptors (1/100, rabbit polyclonal to human TPA1, TRPV1 and TRPM8, all purchased from Abcam, UK), anti-HRV mAb (1/100; QED Bioscience, USA), chicken polyclonal anti-MAP-2 (1/500; Novus Biologicals, USA), anti-ICAM1 (1/100; Abcam, UK) and secondary antibodies anti-rabbit/mouse labelled with Alexa Fluor 488/568 (1:500; Life Technologies, USA) or rabbit anti-chicken (1:50; Jackson ImmunoResearch Laboratory, USA). Detailed methods for both IF and flow cytometry are given in the online supplement.
Standard and qRT-PCR
Primers used for standard HRV RT-PCR were designed to be specific for the negative strand replication intermediate. RNA was extracted using an RNeasy mini kit (Qiagen, UK) according to the manufacturer's instructions. The two-step RT-PCR was carried out using a Verso RT-PCR kit (Thermo scientific, USA). Quantitative (q)RT-PCR was carried out using a QuantiTect Reverse Transcription Kit (Qiagen, UK) for cDNA production and analysed using a Mx3005P qRT-PCR machine (Agilent Technologies, USA). Details of standard and qRT-PCR primers and reaction conditions are provided in the online supplement and in the paper by Galbraith et al.25
Human magnetic bead panel
Supernatants obtained from infected Wi-38 and dIMR-32 cells were freshly prepared for multiplex assay for protein biomarkers using a human magnetic bead panel (Millipore, Germany). Candidate factors IL-1β, IL-6, IL-8 and nerve growth factor (NGF) known to be produced during HRV infection8 ,26 ,27 and associated with neural sensitisation28 ,29 were examined according to the manufacturer's instructions by using Luminex 200 (Millipore, Germany). HRV stock and pelleted virus preparations were similarly examined.
Statistical analysis
All values are presented as a mean of at least three independent experiments. Statistical analysis was performed using one-way analysis of variance followed by the Tukey post hoc test for multiple comparisons of groups. Differences were considered significant at p<0.05.
Results
Differentiated IMR-32 cells express TRP receptor channels
Non-differentiated IMR-32 cells are shown in figure 1A. Following 8-day differentiation, cells acquired the neuronal phenotype including outgrowth of neuronal processes (figure 1B). Staining for the neuron-specific protein microtubule-associated molecule MAP-2 was demonstrated in dIMR-32 cells (figure 1C). The cells also stained for TRPA1, TRPV1 and TRPM8 on cell bodies and along neuronal processes on the surface of non-permeabilised cells (figure 1D–F). Positive control staining on HEK cells transfected with TRPA1, TRPV1 and TRPM8 in non-permeabilised cells are shown (figure 1G–I).

Neuronal-like cultures generated from IMR-32 neuroblastoma cells are positive for transient receptor potential (TRP) channel expression. Phase-contrast images of IMR-32 cultures (A) before and (B) 8 days after BrdU treatment. Arrow indicates cell processes. (C) dIMR-32 cells are positive for neuronal specific marker (MAP-2) staining (red). TRP channel staining (green) was detected on the cell body and along the neuronal processes in dIMR-32 cells (D–F) and stably transfected non-permeabilised (G) HEK-TRPA1, (H) TRPV1 and (I) TRPM8 which were used as positive controls. Nuclei stained with 4′,6-diamidino-2-phenylindole (magnification ×200 for A and B, ×400 for C–I).
Differentiated IMR-32 cells are susceptible to HRV infection
The dIMR-32 and Wi-38 cells (as control) were inoculated with HRV at a MOI of 1. Cytopathic effect was observed in Wi-38 cells at 48 h post infection (hpi) but not in dIMR-32 cultures (figure 2A). We demonstrated the expression of ICAM-1 protein, which is a cell entry receptor for HRV on both cell types (figure 2B). To confirm that viral replication was taking place in dIMR-32 cells, RT-PCR for the virus-negative strand replication intermediate was carried out on RNA extracted from infected and non-infected cells. RNA from infected Wi-38 cells acted as a positive control (figure 2C). Further evidence of HRV infection in dIMR-32 cells was confirmed with the demonstration of viral protein at 48 hpi throughout the cytoplasm and cell processes of the neurons. Staining for HRV in the infected Wi-38 cells was also extensive and accompanied by cell destruction (figure 2D). Quantitative measurements of virus replication were obtained by qRT-PCR. The copy number in Wi-38 cells increased by approximately 1.5 log over 48 h with an approximate 0.5 log increase over this period in dIMR-32 cells (figure 2E).

dIMR-32 cells are susceptible to human rhinovirus (HRV) infection. (A) Phase-contrast images of mock and HRV-infected dIMR-32 (magnification ×200) and Wi-38 cells (magnification ×400) at multiplicity of infection (MOI) of 1 at 48 hpi. Unlike Wi-38 cells, HRV infection on dIMR-32 cells results in no cytopathic effect (CPE). Arrow indicates CPE. (B) Intercellular adhesion molecule 1 expression on dIMR-32 and Wi-38 cells (red). Nuclei stained with 4′,6-diamidino-2-phenylindole (magnification ×400). (C) RT-PCR for negative strand HRV replication intermediate RNA; lanes 1 and 4: mock-infected, lanes 2 and 5: infected at 48 hpi, lanes 3 and 6: treated with UV-inactivated HRV (UV-HRV) for 48 h. RT-PCR was carried out using primers designed to detect the negative sense replicative HRV intermediate (avoids detection of input virus containing genomic positive strand). Primers for β-actin (house-keeping gene) mRNA were used as a positive control. (D) HRV infection at MOI of 1 at 48 hpi on dIMR-32 and Wi-38 cells stained with anti-HRV (green). Nuclei stained with 4′,6-diamidino-2-phenylindole (magnification ×630 for dIMR-32 cells and ×400 for Wi-38 cells). (E) Absolute copy number of HRV was measured by qRT-PCR following infection (MOI of 1) of dIMR-32 and Wi-38 cells at 2, 8, 24 and 48 hpi. (F) Level of nerve growth factor (NGF), interleukin (IL)-1β, IL-6 and IL-8 release by infected dIMR-32 5, 24 and 48 hpi following infection by HRV at MOI of 1 was measured by bioplex analysis. Data plotted as levels above the baseline level of soluble factor released in mock infected cells.
In order to determine whether rhinovirus infection of our neuronal cells induced release of proinflammatory mediators, we examined supernatant from infected dIMR-32 cells at 5, 24 and 48 hpi (figure 2F). Compared with conditioned medium from mock infected dIMR-32 cells, IL-6 levels were significantly increased at 5 hpi (p=0.03) but not at later time points. NGF levels were significantly increased (p=0.03), but only at later time points (48 hpi). No significant increases in IL-1β or IL-8 levels were observed.
Rhinovirus-induced TRP channel upregulation on dIMR-32 cells is dependent on virus titre and time after infection
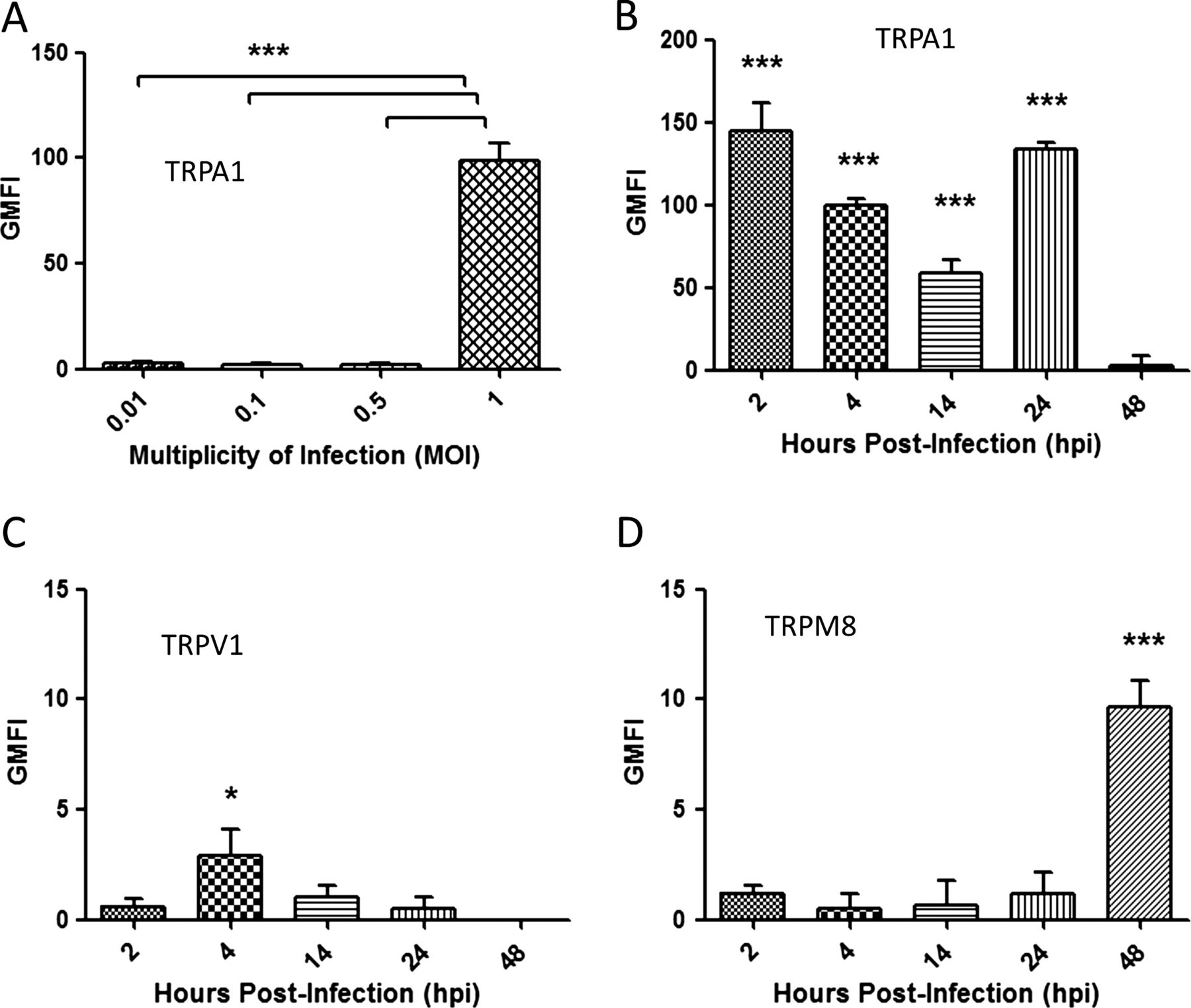
We performed preliminary experiments to determine if TRP channel upregulation on dIMR-32 cells was related to the level of virus infection. The dIMR-32 cells were infected at 0.01, 0.1, 0.5 and 1 MOI and expression measured by FACS analysis at 24 hpi. HEK-transfected cells were used as a positive control for TRPA1, TRPV1 and TRPM8 expression (see online supplementary figure S1A–C). Maximal expression for each TRP channel was noted at 1 MOI (figure 3A and online supplementary figure S1A–C), which was chosen as the optimal MOI for all further studies.

Transient receptor potential (TRP) expression is dependent on multiplicity of infection (MOI) and time after infection. TRP expression was determined by FACS analysis and the levels were quantified by geometric mean fluorescent intensity (GMFI). All graphs were plotted above the TRP baseline level in mock infected cells. (A) The cells were infected at 24 hpi using MOIs of 0.01, 0.1, 0.5 and 1 and stained for TRPA1. dIMR-32 cells were infected at MOI of 1 and stained at 2, 4, 14, 24 and 48 for (B) TRPA1, (C) TRPV1 and (D) TRPM8. Bars represent SD; *p<0.05.
HRV infection significantly upregulated TRPA1 expression at all post infection time points examined compared with mock infected cells, with the highest increase in geometric mean fluorescent intensity (GMFI) levels observed at 2 hpi (145.1±13, p<0.001) and 24 hpi (133.6±3.1, p<0.001) (figure 3B and online supplementary figure S2A, D). In contrast, maximal upregulation of TRPV1 (2.6±1, p<0.05) was observed at the earliest time point only (figure 3 and online supplementay figure S2B,E) whereas maximal upregulation of TRPM8 was only observed at 48 hpi (9.64±1.2, p<0.001; figure 3D and online supplementay figure S2C). Findings at the protein level were matched by measurements of mRNA levels (see online supplementary figure S2F–H). Based on these results, the optimal time points for upregulation of TRPA1, TRPV1 and TRPM8 were selected for all further studies.
Virus-induced soluble factors alone can upregulate mRNA and protein expression of TRPA1 and TRPV1 in dIMR-32 cells
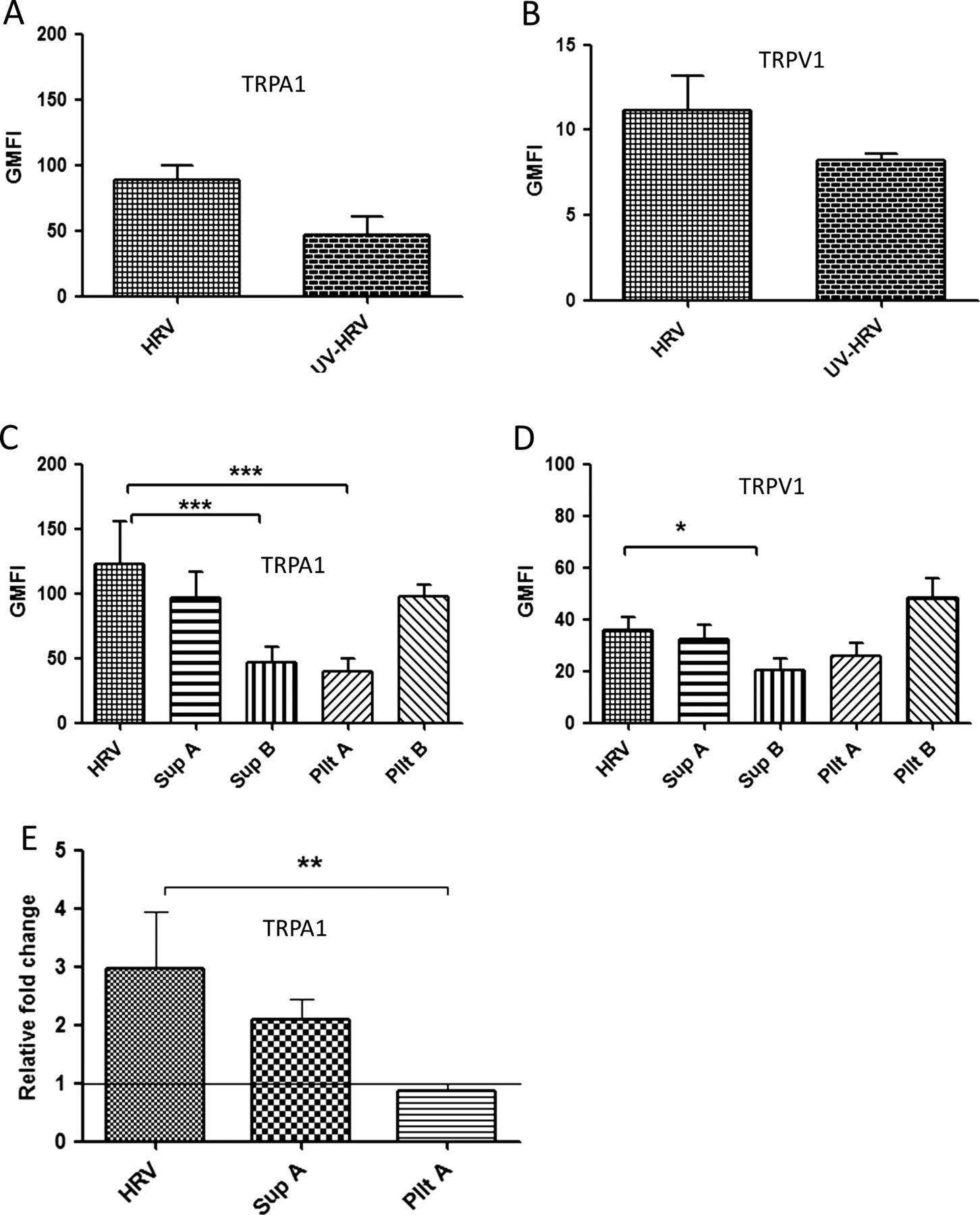
We sought to determine if upregulation of TRP channels following virus infection was due to virus attachment to the cell surface/cell entry, virus replication within the same cell and/or virus-induced factors. TRPA1 was upregulated at 24 hpi by both live virus infection (89±10.3) and, to a lesser extent (46.4±14.6, p=0.1), by the UV-inactivated virus preparation (figure 4A and online supplementary figure S3A). TRPV1 was similarly upregulated, although at an earlier time point (4 hpi), by both live virus infection (11.2±1.9) and the UV-inactivated virus preparation (8.2±0.3, p=0.064; figure 4B and online supplementary figure S3B). As our findings suggested TRPA1 and TRPV1 upregulation was not solely dependent on replicating virus, we wished to determine whether this was due to binding/entry of non-replicating virus particles in cells and/or the presence of virus-induced soluble factors in the inoculum. To establish this, cells were either treated with supernatant generated from Wi-38 cells infected with HRV for 48 hpi at MOI of 1 and ultracentrifuged to remove virus particles (Sup A), supernatant from mock infected Wi-38 cells (Sup B), UV-inactivated pelleted virus (Pllt A) or non-UV inactivated pelleted virus (Pllt B). Standard HRV infection using live virus at an MOI of 1 was used as a positive control (HRV). Standard virus which had been UV-inactivated (UV-HRV) was also included as in the previous experiment.

TRPA1 and TRPV1 expression is upregulated by virus-induced soluble factors. The cells were either infected at a multiplicity of infection of 1 (human rhinovirus (HRV)) or treated with UV-inactivated HRV (UV-HRV), virus-free supernatant (Sup A), supernatant from uninfected Wi-38 cells (Sup B), UV-inactivated pelleted virus (Pllt A) or non-UV inactivated pelleted virus (Pllt B). TRP protein levels were measured by flow cytometry and presented by geometric mean fluorescent intensity (GMFI) and mRNA levels were quantified by qRT-PCR. (A) TRPA1 GMFI at 24 hpi and (B) TRPV1 GMFI at 4 hpi following infection or treatment with UV-inactivated standard virus. (C) TRPA1 GMFI at 24 hpi following infection or treatment with supernatant or pelleted virus preparations. (D) TRPV1 GMFI at 4 hpi following infection or treatment with supernatant or pelleted virus preparations. The graphs were plotted above the TRP baseline level (control–uninfected/untreated stained). (E) TRPA1 mRNA levels after treatments as in (C). Data are presented as relative fold change to TRP levels in uninfected/untreated control cultures. Bars represent SD; *p<0.05, **p<0.01 very significant and ***p<0.001 highly significant. TRP, transient receptor potential.
Treating the cells with supernatant derived from infected dIMR-32 cells gave no response (see online supplementary figure S4). However, we observed upregulation of TRPA1 in dIMR-32 cultures treated with Sup A (prepared from Wi-38 cells), which was of similar magnitude to that seen in cultures infected with live pelleted virus (Pllt B) and with the positive control (figure 4C and online supplementary figure S5A). In contrast, only marginal TRPA1 upregulation was seen following treatment with either mock supernatant (Sup B) or UV-inactivated virus pellet (Pllt A). There was approximately a 50-fold increase in GMFI induced by Sup A over Sup B. Overall, these results suggested that neither direct contact of non-infectious (inactivated) virus particles with dIMR32 cells nor the constitutive soluble factors in mock infected Wi-38 cells effectively upregulate TRPA1. Furthermore, overall our findings suggest that soluble factors released during HRV replication in the Wi-38 lung cells were responsible for upregulation of TRPA1 on dIMR-32 neuronal cells. We observed a similar (although quantitatively lower) pattern of TRPV1 upregulation on dIMR32 cells with approximately a 15-fold increase induced by Sup A over Sup B (figure 4D and online supplementary figure S5B). The findings for protein expression were replicated at the mRNA level (figure 4E).
Virus replication in dIMR32 cells is required for TRPM8 upregulation
In contrast to TRPA1 and TRPV1, the expression of TRPM8 was only upregulated by either live standard HRV or live pelleted virus preparation (Pllt B). The latter induced a slightly higher level of TRPM8 compared with the standard virus (p=0.057; figure 5A). A similar pattern of upregulation at the mRNA level was observed (figure 5B). Our findings indicate that neither attachment/binding/entry of the virus to the cell or the presence of virus-induced soluble factors is sufficient to upregulate TRPM8 but, rather, virus replication within the neuronal cell is necessary.
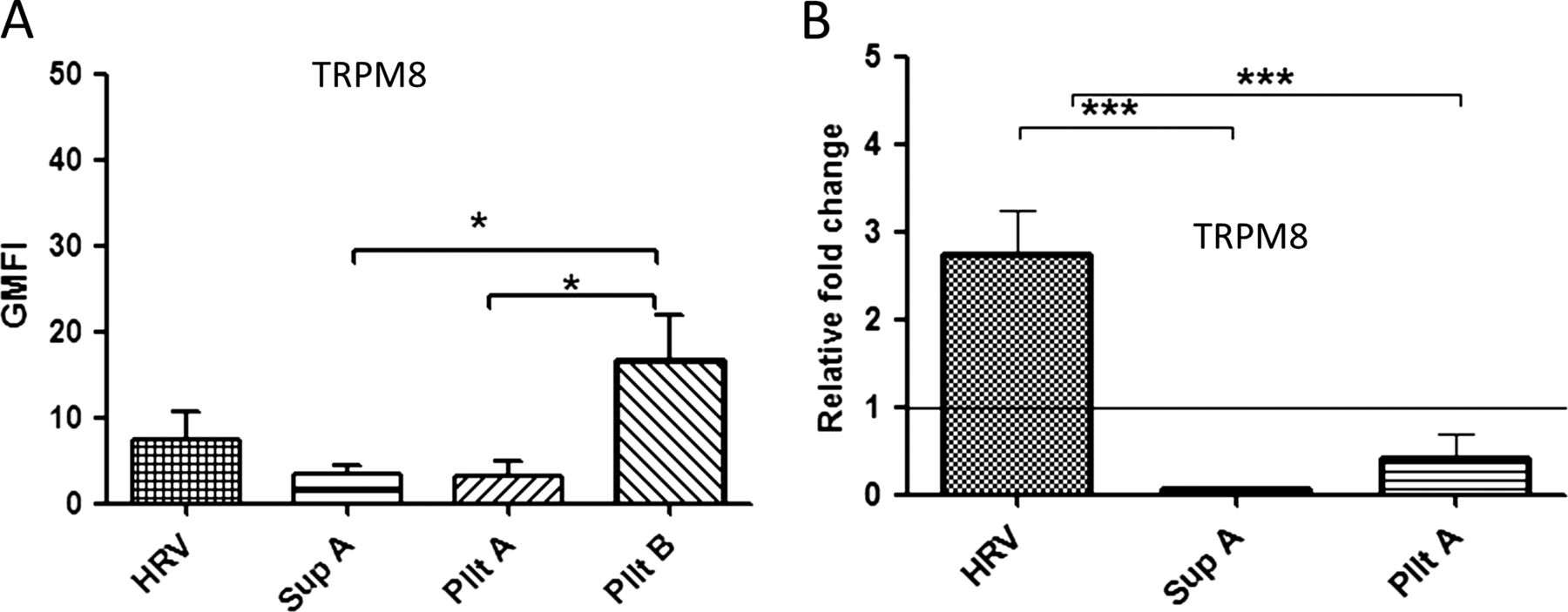
The upregulation of TRPM8 expression is dependent on virus replication. The cells were either infected at a multiplicity of infection of 1 (human rhinovirus (HRV)) or treated with virus-free supernatant (Sup A), inactivated pelleted virus (Pllt A) or non-UV inactivated pelleted virus (Pllt B). (A) TRP protein levels were measured at 24 hpi by flow cytometry and presented by geometric mean fluorescent intensity (GMFI). The graph was plotted above TRP baseline level (control–uninfected/untreated stained) and (B) TRPM8 mRNA levels. Data are presented as relative fold change to TRP in uninfected/untreated control cultures. Bars represent SD; *p<0.05 and ***p<0.001 highly significant. TRP, transient receptor potential.
Bioplex analysis of soluble factors released from HRV-infected Wi-38 lung cells
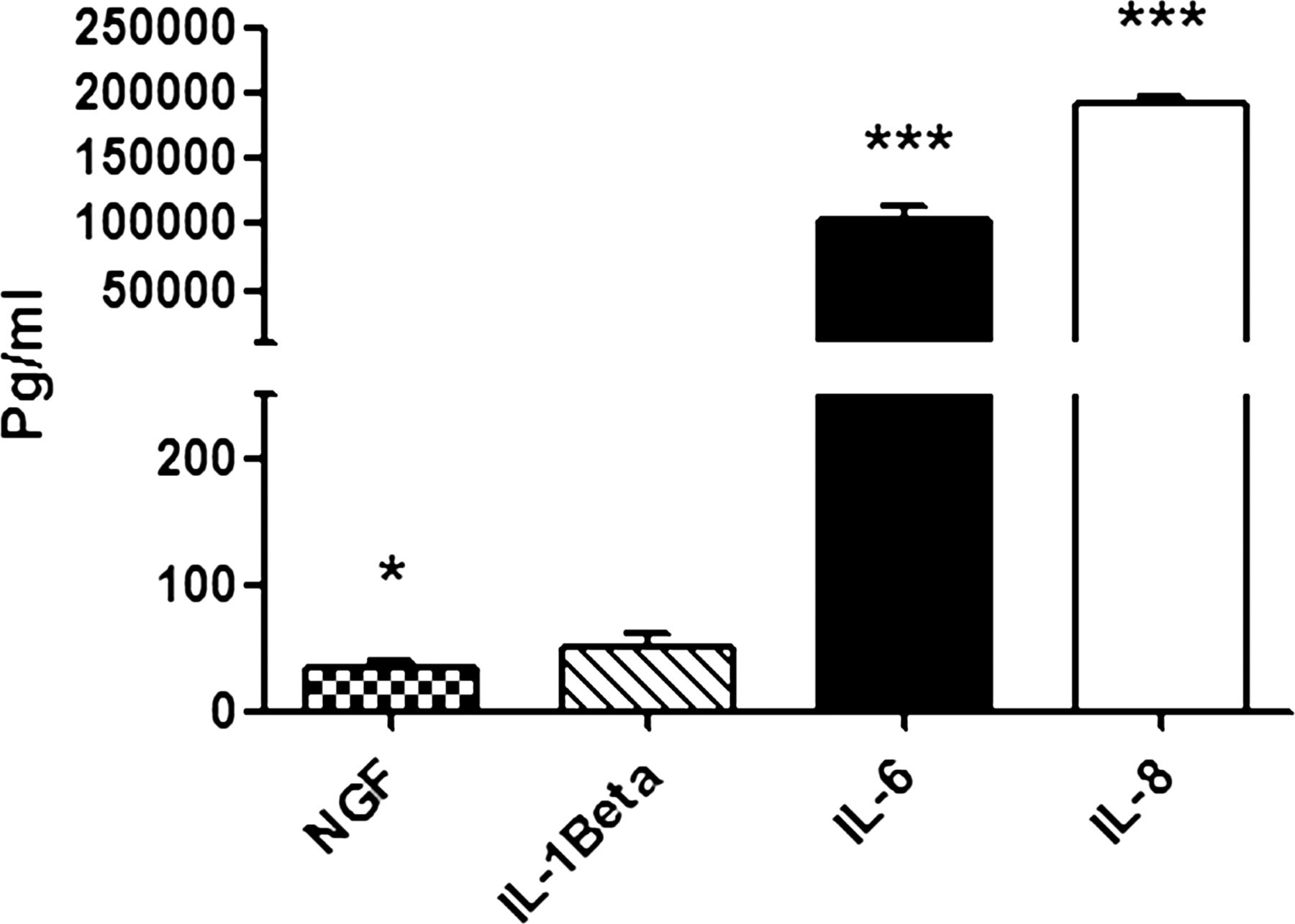
To determine which factors in the supernatant from the Wi-38 lung cells might be responsible for TRPA1 and TRPV1 upregulation, we carried out bioplex analysis of the mediators IL-1β, IL-6, IL-8 and NGF which are known to be induced by rhinovirus infection of epithelial cells (figure 6).8 ,26 ,27 Compared with conditioned medium from mock infected Wi-38 cells (Sup B), the virus supernatant (Sup A) contained significantly increased levels of NGF (p=0.012), IL-6 (p<0.001) and IL-8 (p<0.001) but not IL-1β. To determine that centrifugation was effective in removing soluble factors, we compared levels of the most highly expressed cytokines IL-6 and IL-8 in SupA, Sup B and Pllt B preparations. No significant levels of these cytokines were detected in the virus pellet preparations (see online supplementary figure S6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of nerve growth factor (NGF), interleukin (IL)-1β, IL-6 and IL-8 released by infected Wi-38 cells compared with mock infected cells. The level of soluble factors was measured in supernatant (Sup A) produced from Wi-38 cells. Bars represent SD; *p<0.05, ***p<0.001 highly significant.
Discussion
In this study we have shown for the first time that differentiated human neuronal cells used as a model of sensory neurons can be infected by rhinovirus. Furthermore, we describe the novel finding that incubation with HRV differentially upregulates TRP channel protein expression. We observed early upregulation of TRPA1 and TRPV1 at 2 and 4 hpi, respectively, by a mechanism that appears dependent on virus-induced soluble factors rather than replicating virus. Levels of TRPA1 but not TRPV1 remained elevated for 24 hpi. By contrast, maximal upregulation of TRPM8 was observed at 48 hpi, but only in the presence of replicating virus.
Following a respiratory viral infection, the airways become hypersensitive, manifest clinically with troublesome bouts of cough, which are difficult to treat and associated with impaired health status.30 Almost 50 years ago, Empey and colleagues proposed a mechanism for viral-induced cough and bronchial hyper-responsiveness whereby epithelial damage exposes and thus ‘sensitises’ airway sensory nerves to inhaled irritants.31 The precise mechanism of this sensitisation has yet to be established. Experimental rhinovirus infection in animals induces cough reflex and bronchial hyper-reactivity and within humans following naturally occurring infection heightened cough response to chemical6 and mechanical stimulation,32 is observed.
HRV infects the lower airways, directly triggering the synthesis of inflammatory cytokines from a range of airway cells including epithelial cells, smooth muscle cells, macrophages and fibroblasts.8 ,26 ,27 ,33 To date there has been little attention given to how virus infection alters sensory nerve function. We hypothesised that virus-associated sensory hyper-responsiveness arises at least in part due to upregulation of irritant receptors on sensory nerves. We chose to study three members of the TRP channel family of proteins (TRPV1, TRPA1 and TRPM8) expressed on sensory nerves and other airway cell types. These have been identified as important in airway sensation and nociceptive response to physical and chemical triggers in the environment.12 ,34 We used an immortalised neuroblastoma cell line (IMR-32) and differentiated these cells to model human neurons. The dIMR-32 cells are known to express functional TRP channels.21 In the current study we demonstrated for the first time that these cells are susceptible to rhinovirus infection and represent a suitable model for the study of HRV-induced TRP channel modulation.
Our findings indicate that HRV causes TRP channel upregulation, although differences in the time points for optimal upregulation of specific receptors and the factors responsible were observed. TRPA1 is upregulated early (2 h post infection) and receptor expression remains elevated up to 24 h. We also observed early upregulation of TRPV1 (4 h post infection), but this was to a much lower level and was not evident at later time points. TRPA1 and TRPV1 channels are both considered to have an important role in the regulation of irritant-induced airway responses, in particular cough.17 In an in vivo conscious guinea pig cough model, Grace and colleagues observed that TRPV1 and TRPA1 antagonists each caused partial inhibition of the tussive response to prostaglandin E2 (PGE2) and bradykinin whereas both together rendered complete inhibition.14 The neuronal upregulation of TRPV1 and TRPA1 in response to rhinovirus infection may therefore represent one mechanism responsible for acute viral cough. Upregulation of both these channels also occurred independently of replicating virus as both the UV-inactivated virus preparation and the supernatant from HRV-infected Wi-38 lung fibroblasts (containing virus-induced soluble factors alone) was sufficient to induce increased channel expression. We also showed that NGF, IL-6 and IL-8 represent possible candidates for the soluble factors responsible for upregulation of TRPA1 and TRPV1. Therefore, production of soluble factors from infected epithelium, fibroblasts and other cell types may play an important role in the sensitisation of airway nerves. Elevated levels of NGF and IL-6 were also induced by direct infection of dIMR-32 cells, but at much lower quantities. NGF has recently been shown to be induced by HRV infection of airway epithelial cells and to cause upregulation of ICAM-1 with consequent increase in virus replication.35 We suggest that rhinovirus infection of non-neuronal and neuronal cells in the airways may induce factors which upregulate expression of TRP channels on airway sensory nerves. This will require further investigation.
In contrast to TRPA1 and TRPV1, infection with live standard or live virus derived from the pellet was necessary to upregulate mRNA and protein levels of TRPM8. Neither UV-inactivated virus nor virus-generated soluble factors alone had any effect. These findings suggest that virus undergoes replication to induce TRPM8 mRNA and protein expression in neuronal cells. This is consistent with our observation that upregulation of TRPM8 occurred later at 48 hpi when a higher percentage of cells are infected. It is likely that an intracellular signalling pathway needs to be activated during virus replication, perhaps by a non-structural viral protein or viral RNA/replication intermediate, to upregulate TRPM8.
To our knowledge this is the first study to investigate HRV infection in neuronal cells and to explore the association between TRP channels and virus infection. We have shown that there are at least two mechanisms whereby viruses can upregulate TRP channels and that this is channel-specific. The role of particular factors—NGF, IL-6 and IL-8 (and possibly others)—in TRPAI and TRPV1 upregulation requires further study. Regulation of intracellular pathways in the TRPM8 receptor response is also an area for future detailed investigation. Although our initial findings indicate that TRP channels themselves can be directly targeted therapeutically, the upstream events which induce these receptors during virus infection may allow more novel approaches for the treatment of virus-induced cough.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
SLC and LPAMcG are joint principal investigators.
-
Contributors HA carried out the laboratory work and analysis and helped to draft the paper. SLC is a joint principal investigator/senior author and expert in virology and directed the virological molecular aspects of the work. She also revised drafts of the manuscript. LPAMcG is a joint principal investigator/senior author and expert on cough and the TRP channels. He had input on this aspect with regard to design and interpretation of experiments and also revised drafts of the manuscript. LGH is an expert on asthma and contributed to the experimental approach, interpretation and comments on the manuscript.
-
Funding The work was supported by Asthma UK Foundation grant number 09/017.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.