Article Text

Abstract
Objectives The aim of this study was to test the effectiveness of Provent, an expiratory nasal resistance valve, to prevent the recurrence of OSA following CPAP withdrawal.
Design Randomised, partially blinded, parallel, placebo-controlled trial.
Setting Outpatient sleep clinics in the UK (Oxford) and Switzerland (Zurich).
Participants 67 patients with OSA receiving CPAP were randomised to one of three groups for 2 weeks: continuing CPAP, Provent or placebo Provent.
Main outcome measures Primary outcomes included for Provent versus placebo Provent, OSA severity (oxygen desaturation index (ODI), apnoea–hypopnoea index (AHI)) and Epworth Sleepiness Scale (ESS) score. Secondary outcomes for Provent versus placebo Provent included ODI from ambulatory pulse oximetry and blood pressure (BP). For CPAP versus Provent, or CPAP versus placebo Provent, secondary outcomes included ODI/AHI, ESS and BP.
Results 63 patients were included in the per protocol analysis. OSA recurred in the Provent (ODI 35.8, SD 17.4) and placebo Provent (ODI 28.2, SD 18.3) groups, and there was no significant difference in ODI, AHI and ESS between Provent and placebo Provent at 2 weeks (mean difference ODI −1.0, 95% CI −10.0 to +12.0, p=0.85; AHI +3.2, 95% CI −7.7 to +14.1, p=0.52; and ESS −1.4, 95% CI −4.1 to +1.4, p=0.33). ODI from ambulatory pulse-oximetry and BP at 2 weeks were not different in the Provent versus placebo Provent groups. ODI, AHI and BP, but not ESS, were significantly higher in the Provent and placebo Provent groups compared with CPAP.
Conclusions Provent cannot be recommended as an alternative short-term therapy for patients with moderate to severe OSA already on CPAP.
TrialRegNo NCT01332175.
- Sleep apnoea
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does Provent, an expiratory nasal resistance valve, prevent the recurrence of obstructive sleep apnoea (OSA) following continuous positive airway pressure (CPAP) therapy withdrawal?
What is the bottom line?
-
CPAP therapy is not always tolerated by patients with OSA. Thus there is a need for alternative therapies, particularly for short periods off CPAP.
Why read on?
-
The findings of this randomised controlled trial show that Provent cannot be recommended as an alternative short-term therapy for patients with moderate to severe OSA already on CPAP.
Introduction
Obstructive sleep apnoea (OSA) is a common sleep-related breathing disorder affecting up to 30% of middle-aged adults in Western countries.1 OSA is characterised by transient interruption of ventilation during sleep caused by complete or partial collapse of the upper airway, resulting in apnoeas and hypopnoeas. These interruptions of ventilation lead to oxygen desaturation, increased inspiratory effort, arousal from sleep and, as a consequence, increased daytime sleepiness, impaired quality of life, increased risk of traffic accidents and hypertension.2–4
Several therapies have been developed to treat OSA, such as continuous positive airway pressure (CPAP), oral appliances, and upper airway surgery. CPAP is the most effective treatment to prevent the repetitive upper airway obstruction and to improve sleepiness and quality of life. However, adherence to treatment is an important factor limiting the effectiveness of CPAP therapy.5 Moreover, CPAP utilisation may be difficult while travelling or on holiday due to absence of electricity or due to the cumbersome size and weight of the equipment. Therefore, alternative therapies which could replace CPAP for short or longer periods are needed.
A nasal expiratory positive airway pressure device, Provent (Ventus Medical, Belmont, California, USA) has been proposed as a new therapeutic option for patients with OSA who cannot tolerate CPAP therapy or are not compliant with it. This device consists of small disposable one-way valves applied to both nares by adhesive tape. The valves have negligible inspiratory resistance, but offer resistance during the expiratory phase, thus creating a positive end-expiratory pressure, thereby increasing end-expiratory lung volumes.6 This is thought to traction the upper airways, decreasing pharyngeal wall compliance, thus preventing them from collapsing.6 There may be an additional effect from the higher pharyngeal pressures during expiration, perhaps carrying over into inspiration, thus splinting open the pharynx; however this would be expected to disappear as inspiration begins. Previous pilot studies, and one randomised controlled trial, have suggested that Provent might be effective in reducing sleep-disordered breathing and daytime sleepiness in patients with mild to moderate OSA, however the individual patient responses were considerably variable.7–9
The aim of the current study was to investigate the effectiveness of Provent on sleep-disordered breathing and daytime symptoms in patients with moderate to severe OSA who had already been successfully treated with CPAP during a 2-week period of CPAP withdrawal. If effective, this would be useful to cover a short period off CPAP, such as during a holiday.
Methods
Patients
Patients previously diagnosed with OSA and treated with CPAP who were registered in a database of the Sleep Disorders Centre and Pulmonary Division of the University Hospital Zurich, Zurich, Switzerland, and of the Centre for Respiratory Medicine, Oxford, UK were eligible for the trial if they fulfilled the following criteria: aged between 20 and 75 years, had an oxygen desaturation index (ODI, ≥4% dips) of >10/h during their original diagnostic sleep study, an ODI >10/h during an ambulatory nocturnal pulse oximetry performed on the last night of a four-night period without CPAP prior to the main study, and if they had been treated with CPAP for >12 months with an average compliance of ≥4 h per night.
Patients with previous ventilatory failure, Cheyne–Stokes breathing, unstable and untreated coronary or peripheral artery disease, severe and inadequately controlled arterial hypertension, a history of any sleep-related accident, or were current professional drivers were excluded from the study.
Recruitment of patients started in April 2011 and the trial was completed in August 2012. The trial was conducted according to the Declaration of Helsinki and was approved by the research ethics committees in both Zurich and Oxford (EK-1600, 11/NW/0370 respectively) and registered (NCT 01332175). Written informed consent was obtained from all participants.
Trial design, randomisation and interventions
This was a randomised, dual-centre, parallel, placebo-controlled trial. Blinding of the patients was possible as to which Provent device they received, but clearly not between CPAP and the two Provent devices. Those performing the analysis were not involved in the day-to-day running of the trial and were blind to the treatment allocation. After confirming the persistence of OSA by home overnight pulse oximetry (Pulsox-300i, Minolta, Osaka, Japan) on the last night of a four-night period without CPAP, eligible patients returned to therapy with CPAP for ≥14 days. After baseline assessments, patients were randomised by a central telephone service using a computer-derived trial number allocation. The randomisation incorporated minimisation using the following four factors: sleep apnoea severity from the original sleep study (≥4%SaO2 dips/h, ≥25 or <25 dips/h), body mass index (≥33 or <33 kg/m2), current Epworth Sleepiness Scale (ESS) score (≥7 or <7), and participating centre (Minim, http://www-users.york.ac.uk/~mb55/guide/minim.htm). Patients were allocated equally to one of three groups: continue with autoCPAP therapy (ResMed, S8) or switch to either Provent or placebo Provent for 2 weeks. Provent and placebo Provent devices were supplied by the company in sealed and colour-coded bags. The placebo devices were identical in all respects except that the valve membranes had been removed, allowing unobstructed expiratory flow and unobstructed inspiratory flow. Patients were instructed by a trainer on how to apply the Provent devices and visually verified proper application; this was done without describing to the subject the mechanisms of action of the device, which might have allowed them to infer their group allocation. After 2 weeks, follow-up assessments were performed.
Sample size estimation
The sample size was estimated on the assumption that a minimally important difference in the ODI between Provent and placebo Provent would be 10/h (SD 10).10 Based on this assumption, a power calculation indicated that 20 patients would be required to complete the trial in each arm to ensure this minimally important difference in ODI was not missed with a power of 87%. If the minimally important difference in the ESS between Provent and placebo Provent was set at 2.5 points (SD 2.5), as found in our previous 2-week CPAP withdrawal study,10 then a power calculation indicated that 20 patients were also required. Sample size calculations were not specifically required for the apnoea–hypopnea index (AHI) as this is highly related to ODI.
Primary outcome measures
Coprimary outcome measures for Provent versus placebo Provent were OSA severity assessed by ODI/AHI from in-hospital sleep studies, and the ESS.
Secondary outcome measures
Secondary outcome measures for Provent versus placebo Provent included OSA severity assessed by ODI from nightly ambulatory pulse oximetry, and blood pressure. Further secondary outcomes were related to the comparison between CPAP and Provent (or placebo Provent): ODI/AHI from in-hospital sleep studies and ambulatory pulse oximetry, ESS, and blood pressure.
Sleep studies
At baseline, and at 2 weeks, respiratory polysomnographic sleep studies were performed and analysed using automated software and manual checking, according to standard methods. Measurements comprised pulse oximetry, ECG, body position, audio-visual recordings by a low-light infrared video camera. Nasal cannulae were attached to real and sham devices during the second sleep study to analyse nasal flow. Apnoeas were defined as a reduction in amplitude of the nasal flow or chest wall motion by >90% from baseline over the previous 2 min for >10 s; hypopnoeas were defined as a reduction in amplitude of the nasal flow or chest wall motion by >50% from baseline over the previous 2 min for >10 s, associated with a ≥4% drop in oxygen saturation. The severity of sleep-disordered breathing was quantified as the number of apnoeas–hypopnoeas (AHI) and oxygen desaturations ≥4% per hour of study (ODI). In addition, pulse oximetry was performed every night at home during the 2-week study period (Pulsox-300i) to provide serial ODI values.
Subjective sleepiness
Subjective sleepiness was assessed by the ESS at baseline and at 2 weeks.11 ,12
Blood pressure
At baseline and follow-up, blood pressure was measured three times in the morning with a standard digital automatic monitor (Omron Healthcare Co., Kyoto, Japan) in the sitting position after a period of rest of 5 min. The average of these three blood pressure measurements was used for further analysis.
Treatment adherence
Adherence to Provent and placebo Provent was assessed using the diaries kept by the patient during the 2-week experimental period, and expressed as percent nights that the device was worn. Adherence to Provent and placebo Provent during the in-hospital sleep studies was ensured by video. CPAP usage data were downloaded from the machines’ internal memory.
Statistical analysis
An analysis plan was written before the data were analysed. All values are presented as mean (SD) unless otherwise stated. Statistical analyses were performed with SPSS (V.20.0 for Windows). The primary analysis was a per-protocol analysis (use of allocated placebo or Provent devices for >75% of the nights in the assessment phase); a per-protocol analysis was used as we were interested in the physiological effects of Provent, undiluted by potential compliance issues. We compared placebo and Provent devices for OSA severity (as measured by the AHI and ODI), and symptoms (as measured by ESS). Comparison between treatment groups was performed using multivariate regression analysis to calculate the treatment effect, with adjustment for baseline values (table 1) significantly associated with the outcomes (only the ODI during the preliminary four-night CPAP withdrawal reached p<0.1, the level required for inclusion), and neck circumference, which was regarded as of particular biological plausibility and might have biased the result if unbalanced at baseline (whether or not any difference reached p<0.1). Cases in which primary outcome data were missing were not analysed, however a sensitivity analysis was carried out for the primary outcomes under best and worst case scenarios. Nightly ODI from ambulatory pulse-oximetry measurements were compared between the groups by repeated measure analysis of variance. Statistical significance was set at p<0.05, with no correction for having two primary endpoints (OSA severity and ESS), given that they are only minimally related.
Baseline characteristics
Results
Trial profile, patient characteristics and adherence to therapy
The three groups were similar regarding patient characteristics at baseline (table 1). Data on AHI downloaded from the patients’ CPAP machines and ESS values within the normal range proved successful treatment of patients with CPAP prior to the study (table 1). One patient in the Provent group withdrew from the study because of a return of intolerable daytime symptoms. Three patients (one in the Provent group, two in the placebo Provent group) were excluded from the per-protocol analysis because of insufficient compliance. The average nightly compliance in the CPAP group was 418 (SD 81) min. The median percentage of nights Provent was used was 99.0% (85.2%–99.0%) and placebo Provent 100% (81.7%–100%). Figure 1 shows the trial profile and patient flow.

Patient flow. CPAP, continuous positive airway pressure.
Primary outcomes
OSA severity from in-hospital sleep studies
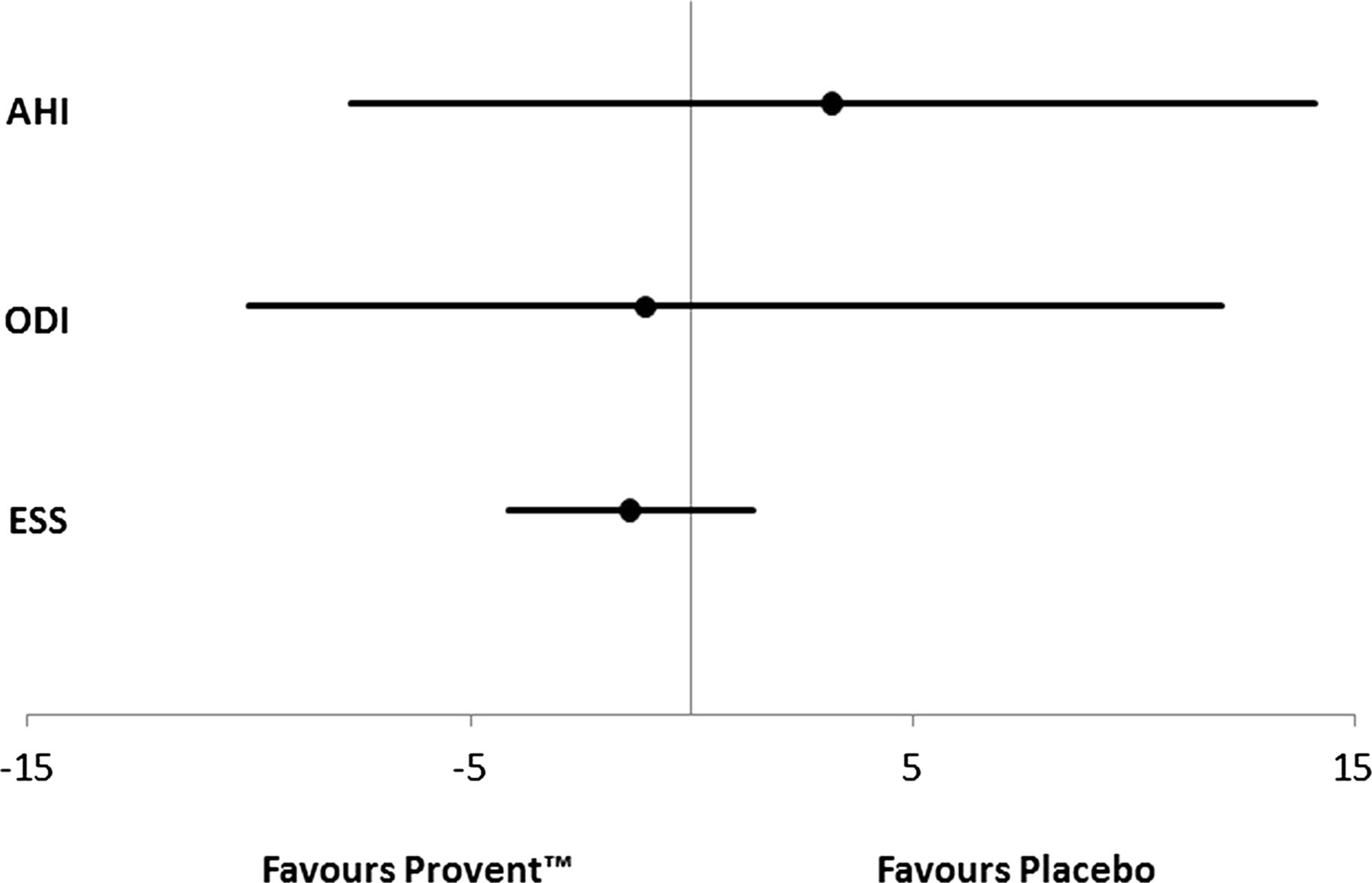
OSA recurred in the Provent and placebo Provent groups, and there was no significant difference in the treatment effect between Provent and placebo Provent groups for measures of OSA severity at 2 weeks (mean difference ODI −1.0, 95% CI −10.0 to +12.0, p=0.85; AHI +3.2, 95% CI −7.7 to +14.1, p=0.52) (figure 2). Sensitivity analysis, for best and worst case scenarios for the single case with missing outcome data, did not materially alter the results.

Forest plot showing no statistically significant treatment effect of Provent on apnoea–hypopnoea index (AHI, events per hour), oxygen desaturation index (ODI, events per hour) and Epworth Sleepiness Scale (ESS) at 2 weeks compared with placebo Provent.
Subjective sleepiness
There was no significant difference in ESS treatment effect between the Provent and placebo Provent groups at 2 weeks (mean difference −1.4, 95% CI −4.1 to +1.4, p=0.33) (figure 2).
Secondary outcomes
OSA severity from nightly home oximetry
OSA recurred in the Provent and placebo Provent groups during the first night off CPAP, and there was no significant difference in ODI derived from home oximetry between Provent and placebo Provent across the 2 weeks (p=0.66) (figure 3).

Oxygen desaturation index (ODI) derived from nightly home pulse oximetry during the study period (solid line: Provent; short-dashed line: placebo Provent; long-dashed line: continuous positive airway pressure (CPAP)). There was no significant difference in ODI between Provent and placebo Provent on any night (p=0.66). ODI was significantly lower in the CPAP group compared with Provent and placebo Provent at any time point (p<0.001).
Blood pressure
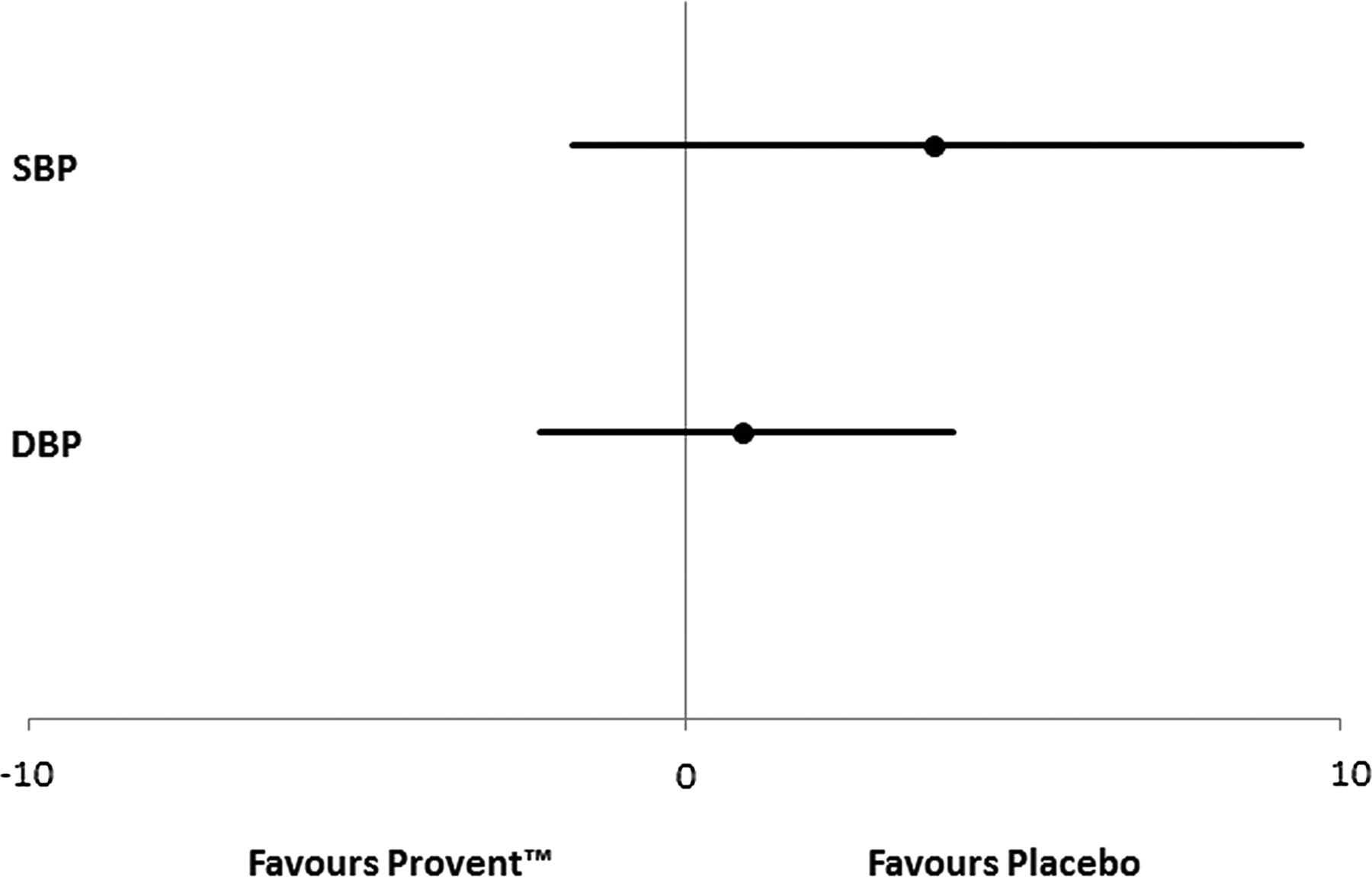
There was no significant difference in the treatment effect on systolic and diastolic blood pressure between the Provent and placebo Provent groups at 2 weeks (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forest plot showing no statistically significant treatment effect of Provent on systolic (SBP, mm Hg) and diastolic (DBP, mm Hg) blood pressure at 2 weeks compared with placebo Provent.
Comparison of CPAP to Provent and placebo Provent
There was a statistically significant difference in the treatment effect on AHI, ODI and diastolic blood pressure between CPAP and Provent and placebo Provent, respectively, at 2 weeks (table 2). Similarly, there was a statistically significant difference in home oximetry derived ODI between CPAP and Provent (p<0.001) and placebo Provent (p<0.001) across the 2 weeks (figure 3).
Comparison of CPAP to Provent and placebo Provent
ESS increased with Provent and placebo Provent vs CPAP at 2 weeks, but this did not reach statistical significance (table 2).
Discussion
The objective of this randomised, placebo-controlled trial was to test the ability of Provent to prevent the recurrence of sleep apnoea following the withdrawal of CPAP in patients with originally moderate to severe OSA, already well established on this therapy. We have been unable to demonstrate a therapeutic effect of Provent versus placebo Provent on sleep-disordered breathing in patients with OSA already on CPAP, who have their treatment withdrawn for 2 weeks.
Despite CPAP being the gold standard for the treatment of OSA, compliance with this therapy is often less than optimal, with a proportion of patients not tolerating it. Even compliant users often temporarily stop their CPAP therapy, for example, during vacations or business trips, which results in an immediate return of OSA.5 ,10 ,13–15 Thus, there is a need for an alternative and well tolerated OSA treatment, especially during short periods off CPAP.
Provent has been suggested to be an effective alternative treatment in patients with newly diagnosed and untreated OSA, and in those not tolerating CPAP.8 ,9 However, so far there are data from only one randomised, placebo-controlled trial investigating the effects of Provent on OSA.9 In the trial by Berry and colleagues9 patients with primarily mild to moderate, untreated OSA were included, and randomly assigned to either Provent or placebo Provent for 3 months. Sleep studies were performed on two non-consecutive nights, with and without the allocated device, in the first week and 3 months after randomisation. The primary endpoint was comparison of the difference in the AHI between device-on and device-off nights, Provent versus placebo Provent, at week 1. There was a reduction in AHI from 13.8 to 5.0/h in the Provent group versus from 11.6 to 11.1/h in the placebo Provent group, and similar reductions in AHI were found at 3 months.9
In contrast to the study by Berry et al9 we were not able to find therapeutic effects of Provent on indices of OSA severity and subjective sleepiness (figure 2), despite excellent reported adherence to Provent and placebo Provent. The ODI derived from nightly home oximetry implied that OSA recurred within the first night off CPAP in the Provent and placebo Provent groups (figure 3). Moreover, the recurrence of OSA was accompanied by an increase in diastolic blood pressure at 2 weeks (table 2), implying that this cardiovascular consequence of OSA cannot be prevented by Provent in patients with OSA who withdraw their CPAP treatment during holidays.
However, there are some differences between our trial and the study by Berry et al,9 which possibly explain some of the different outcomes. Whereas we included patients with OSA who were well established on CPAP therapy for more than 1 year, Berry et al.9 included patients with newly diagnosed, or previously diagnosed but untreated, OSA. In addition, the patients in the current trial had more severe OSA (average original AHI of study population 38/h) compared with the study by Berry et al (median AHI 13.8 and 11.1/h in the Provent and placebo Provent group, respectively). This is explained by the fact that patients with severe night-time oxygen desaturations were specifically excluded from the study by Berry et al,9 and also a substantial number of their patients had an AHI of less than 5/h on device-off nights, suggesting that some of the included patients did not have clinically significant OSA.
The current study has some limitations. Although we did not perform full polysomnography, the in-hospital respiratory polygraphies were performed with video surveillance, thus ensuring adherence to the allocated treatment. In addition, the findings of this study, which specifically selected patients with optimal CPAP compliance, cannot be generalised without caution to patients with less favourable compliance. Only 14% of those eligible agreed to take part in the study, usually because they did not want to go without CPAP for 2 weeks, or were too busy to partake. It is unlikely that those who agreed to take part in the study are in some way physiologically different, but they may not be representative of all CPAP users in other ways.
In conclusion, we have been unable to demonstrate a therapeutic effect of Provent compared with placebo Provent in patients with moderate to severe OSA, already well established on CPAP therapy, who have their treatment withdrawn for 2 weeks. Thus Provent cannot be recommended as an alternative short-term therapy for patients on CPAP with originally moderate to severe OSA.
References
Footnotes
-
Contributors Design of the study: MK, JRS, KEB; acquisition of data: VAR, BW, JF, CFC; data analysis and interpretation: VAR, NMR, LMY, JRS, MK; manuscript draft and revision for intellectual content and approved the final version: VAR, BW, NMR, LMY, JF, CFC, KEB, JRS, MK. Guarantor of the study: MK.
-
Funding This work was supported by Swiss National Science Foundation grant number 32003B_124915.
-
Competing interests All authors have completed the ICMJE uniform disclosure at www.icmje.org/coi_disclosure.pdf and declare the Swiss National Science Foundation (32003B_124915) provided an unrestricted project grant, Ventus Medical made an unconditional contribution to the Respiratory Trials Unit, Oxford University, UK. The funder was not involved in any part of the trial (other than the supply of Provent and placebo devices) or writing of the manuscript.
-
Ethics approval Research ethics committees in Zurich and Oxford (EK-1600, 11/NW/0370 respectively).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The relevant anonymised patient level data are available on reasonable request from the authors.
Linked Articles
- Airwaves