Article Text

Abstract
Background The delivery of antipseudomonal antibiotics by inhalation to Pseudomonas aeruginosa-infected subjects with non-cystic fibrosis (CF) bronchiectasis is a logical extension of treatment strategies successfully developed in CF bronchiectasis. Dual release ciprofloxacin for inhalation (DRCFI) contains liposomal ciprofloxacin, formulated to optimise airway antibiotic delivery.
Methods Phase II, 24-week Australian/New Zealand multicentre, randomised, double-blind, placebo-controlled trial in 42 adult bronchiectasis subjects with ≥2 pulmonary exacerbations in the prior 12 months and ciprofloxacin-sensitive P aeruginosa at screening. Subjects received DRCFI or placebo in three treatment cycles of 28 days on/28 days off. The primary outcome was change in sputum P aeruginosa bacterial density to the end of treatment cycle 1 (day 28), analysed by modified intention to treat (mITT). Key secondary outcomes included safety and time to first pulmonary exacerbation—after reaching the pulmonary exacerbation endpoint subjects discontinued study drug although remained in the study.
Results DRCFI resulted in a mean (SD) 4.2 (3.7) log10 CFU/g reduction in P aeruginosa bacterial density at day 28 (vs −0.08 (3.8) with placebo, p=0.002). DRCFI treatment delayed time to first pulmonary exacerbation (median 134 vs 58 days, p=0.057 mITT, p=0.046 per protocol). DRCFI was well tolerated with a similar incidence of systemic adverse events to the placebo group, but fewer pulmonary adverse events.
Conclusions Once-daily inhaled DRCFI demonstrated potent antipseudomonal microbiological efficacy in adults with non-CF bronchiectasis and ciprofloxacin-sensitive P aeruginosa. In this modest-sized phase II study, DRCFI was also well tolerated and delayed time to first pulmonary exacerbation in the per protocol population.
- Bronchiectasis
- Respiratory Infection
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does the once-daily inhalation of a dual release liposomal ciprofloxacin formulation reduce airway bacterial load in non-cystic fibrosis bronchiectasis subjects with at least one ciprofloxacin-sensitive Pseudomonas aeruginosa bacterial strain, without tolerability issues?
What is the bottom line?
-
Dual release ciprofloxacin for inhalation appeared well tolerated and resulted in large reductions in airway P aeruginosa bacterial load.
Why read on?
-
In the per protocol population, dual release ciprofloxacin for inhalation also delayed time to first pulmonary exacerbation, a key secondary outcome measure.
Introduction
Non-cystic fibrosis (CF) bronchiectasis remains a condition for which there are few therapies of proven benefit and no licensed therapies to date. The need for well-designed randomised-controlled trials to inform clinical management is a priority. Pulmonary Pseudomonas aeruginosa infection in non-CF bronchiectasis is associated with worse quality of life,1 more pulmonary exacerbations2 and more rapid lung function decline.3 The direct delivery of antipseudomonal antibiotics to the respiratory tract of these subjects by inhalation represents a logical treatment paradigm and the proven efficacy of this approach in CF4 provides an appealing blueprint. However, prior attempts to translate proven CF therapies into non-CF bronchiectasis have been unsuccessful to date. Neither recombinant human DNase I nor tobramycin solution for inhalation (TSI/ TOBI) improves clinical outcomes in this patient population.5 ,6
In spite of microbiological efficacy in non-CF bronchiectasis, inhaled TOBI increases respiratory adverse events (AEs).6 This uncoupling of microbiological and clinical efficacy has been reproduced in another randomised-controlled study of TOBI7 and poor tolerability confirmed in other studies of both TOBI8 and alternative tobramycin solutions.9 The poor tolerability of inhaled aminoglycoside antibiotics in non-CF bronchiectasis may relate to high concentrations of antibiotic contacting the airways during inhalation and the better outcomes reported with low dose nebulised gentamicin in a recent study seem to support this.10
Liposomal encapsulation of inhaled antibiotics may improve tolerability without sacrificing microbiological efficacy by minimising the amount of free antibiotic in direct contact with the airway during inhalation and yet still releasing adequate drug dose to the lower airways. Dual release ciprofloxacin for inhalation (DRCFI, Pulmaquin; Aradigm Corporation, Hayward, California, USA) is a mixture of liposomal and free ciprofloxacin. This formulation has been developed with a view to optimising airway delivery characteristics, being distinguished by both an immediate effective dose (free component) and sustained delivery over 24 h (liposome-encapsulated component).
Preclinical studies have confirmed favourable pharmacokinetic characteristics of inhaled liposomal ciprofloxacin. In animal models, the liposomal component has a lung clearance half life after inhalation of approximately 12 h11 (compared with approximately 1 h for free ciprofloxacin)12 supporting once-daily dosing. Additionally, inhalation of liposomal ciprofloxacin has demonstrated superior efficacy to unencapsulated ciprofloxacin in a murine model of lethal pulmonary Francisella Tularensis infection.12 Human studies of DRCFI confirm a systemic half life of approximately 10 h and sputum ciprofloxacin concentrations persistently above 20 μg/g out to 22 h.13 A comprehensive programme of phase I and II studies (comprehensively reviewed in14) suggest optimal pharmacokinetic properties and microbiological efficacy of the DRCFI formulation and dose evaluated in the current study.
The primary objective of ORBIT-2 (Once daily Respiratory Bronchiectasis Inhalation Treatment), a 24-week, phase II efficacy and safety study, was to evaluate the microbiological efficacy of 28 days of inhaled DRCFI. The total study duration of 24 weeks was selected to provide the opportunity to assess safety, tolerability and generate pulmonary exacerbation data. Given the increased risk of respiratory AEs demonstrated previously with inhaled therapies in non-CF bronchiectasis,5–9 subjects reaching the pulmonary exacerbation endpoint were discontinued from further exposure to trial medication (but remained in the study) to mitigate any possibility of a similar experience with DRCFI.
Methods
See the online data supplement for full details relating to all methods.
Subjects
Clinically stable adults with CT scan-proven bronchiectasis, P aeruginosa airway infection and ≥2 pulmonary exacerbations requiring antibiotic therapy in the preceding 12 months were eligible. Subjects with CF, bronchopulmonary aspergillosis or pulmonary non-tuberculous mycobacterial infection were excluded. At least one ciprofloxacin-sensitive P aeruginosa strain needed to be cultured from sputum during a 14-day screening period in order to proceed to randomisation. The study was approved by the ethics committee review at each study site and all subjects provided written consent.
Study design and procedures
This was a 24-week, multicentre, randomised, double-blind, placebo-controlled study of DRCFI undertaken in 11 sites in Australia and New Zealand. Eligible subjects were centrally randomised 1:1, stratified by number of pulmonary exacerbations (2–3 or ≥4) in the preceding 12 months. DRCFI consisted of liposomal ciprofloxacin for inhalation (150 mg in 3 ml) and free ciprofloxacin (60 mg in 3 ml), each provided in separate vials. Matched placebo consisted of control liposomes (15 mg in 3 ml) and normal saline (0.9%, 3 ml). Subjects nebulised DRCFI or matching placebo once-daily through a PARI LC Sprint powered by a PARI Turbo Boy-S compressor (PARI, Richmond, USA) for up to three treatment cycles of 28 days ‘on’ inhaled therapy, 28 days ‘off’ (figure 1; see online data supplement). Trial medication was discontinued once subjects reached the pulmonary exacerbation endpoint although subjects remained in the study and continued trial visits and assessments. Hence, all subjects (excepting subjects who withdrew from the study for AEs prior to experiencing pulmonary exacerbation) contributed to pulmonary exacerbation data.
Subjects were reviewed after 2, 4 and then every 4 weeks. Assessments performed at each visit included spirometry, sputum collection, 6 min walk test (6MWT) and St George's Respiratory Questionnaire (SGRQ). Sputum was collected 24 h postdose at the end of each ‘ON’ period and transferred at ambient temperature15 by courier on the day of collection to a central laboratory (Dorevitch Pathology, Melbourne, Australia). Culture and identification of pathogens, P aeruginosa quantitative bacteriology and ciprofloxacin minimal inhibitory concentration measures of P aeruginosa isolates were performed.15 For quantitative bacteriology, sputum was homogenised 1:1 with Sputasol (Oxoid, Basingstoke, UK) and serially diluted with sterile saline. The 10 μl samples were inoculated onto chocolate agar and incubated for 24 h. The colony forming unit (CFU) count from the lowest dilution plate containing 30–300 discrete colonies was recorded.
Outcomes
The primary efficacy variable was mean change in sputum P aeruginosa bacterial density (as log10 CFU/g of sputum) from baseline to the end of the first treatment cycle (day 28), comparing treatment with placebo and assessed on the full analysis set (all subjects who received at least one dose of study drug, hereafter modified intention to treat, mITT). Secondary outcome measures included time to first pulmonary exacerbation, forced expiratory volume in 1 s (FEV1), 6MWT, SGRQ, safety and tolerability. Protocol-defined pulmonary exacerbation, using a modification of Fuchs criteria,16 was defined as deterioration in at least four of the following nine symptoms or signs: sputum production (volume, colour, consistency or haemoptysis), dyspnoea, cough, fever, wheezing, exercise tolerance (or fatigue/lethargy/ malaise), FEV1 or FVC fall of at least 10%, new changes on chest radiograph and changes in chest sounds on auscultation.
Data analysis
Using estimates of effect derived from pilot data (unpublished), 40 subjects would be needed to demonstrate a 4 log10 CFU/g (SD 3.5) difference between the active and placebo arms, with 90% power at the 0.05 significance level. The primary outcome was assessed on the mITT population by analysis of covariance with effects for annual pulmonary exacerbations as a blocking variable, the baseline value for P aeruginosa bacterial load as a covariate and the treatment effect. Per protocol analyses (defined as all randomised patients who did not experience major protocol deviations) were also performed. Time to pulmonary exacerbation was assessed by Kaplan–Meier survival curves. Given the trial design stipulating withdrawal from study drug following pulmonary exacerbation, data analysis for the majority of outcomes was prespecified on day 28 data although data were collected throughout the duration of the trial.
Results
Between November 2009 and September 2010, 70 subjects were screened and 42 (22 placebo and 20 DRCFI) randomised (see figure 1 for trial flow and table 1 for patient demographics). A single subject was randomised in spite of not culturing P aeruginosa at screening in violation of the protocol. All 42 randomised subjects were included in the mITT analysis, and 39 (93%) completed assessments at the final visit on day 168.
Baseline characteristics of the subjects

Trial flow diagram (AE, adverse event; CFUs, colony forming units; DRCFI, dual release ciprofloxacin for inhalation; mITT, modified intention to treat; PEx, pulmonary exacerbation; after randomisation, n refers to the number of subjects continuing to receive trial medication, although all subjects were encouraged to continue trial assessments until completion at day 168).
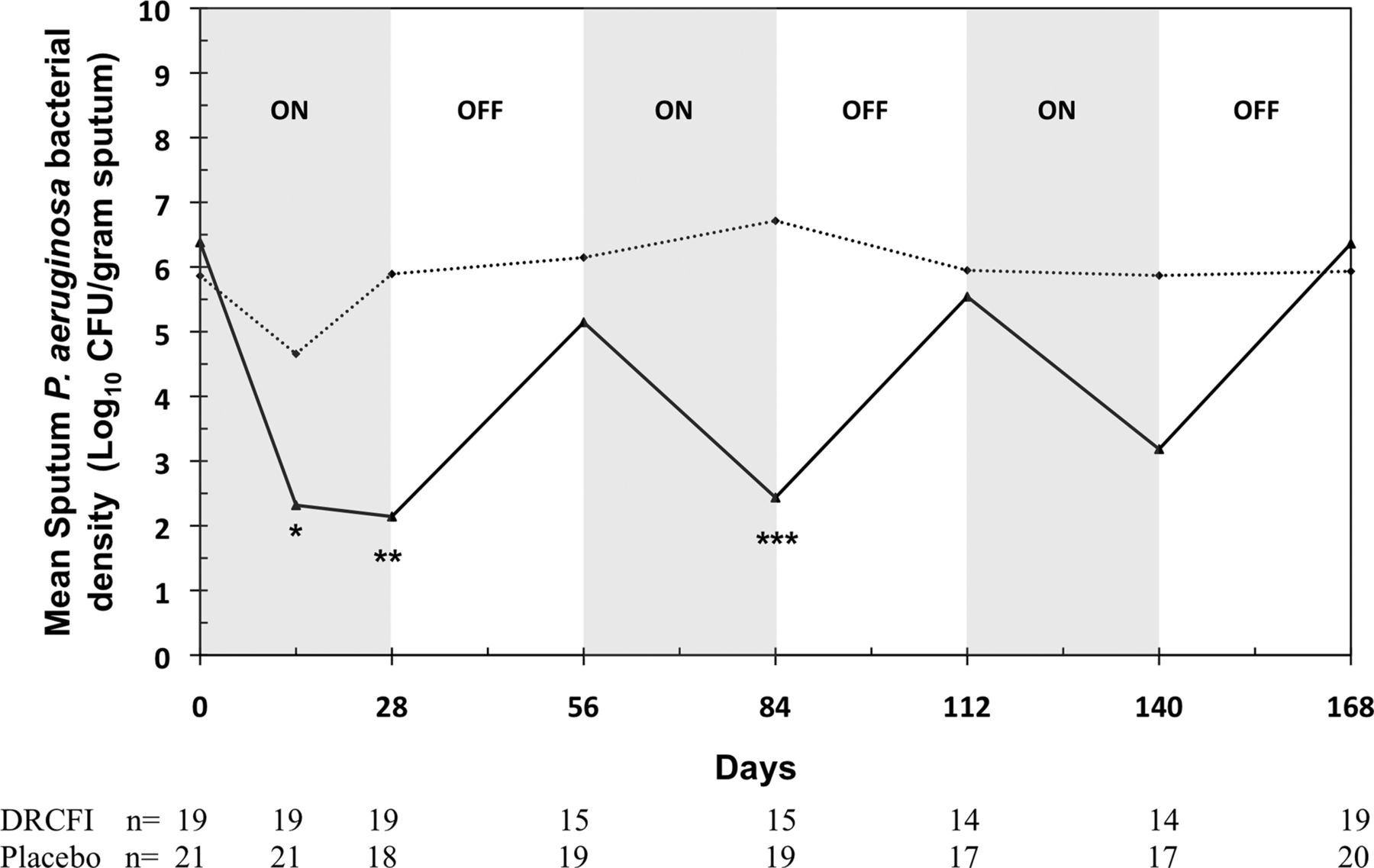
DRCFI resulted in significant reductions in P aeruginosa bacterial density to day 28 compared with placebo (−4.2±3.7 vs −0.08±3.8 log10 CFU/g, p=0.002; figure 2). Limiting the analysis to the per protocol population showed a similar, statistically stronger, treatment effect (p<0.001). The reduction in bacterial density was seen early on, from the first visit at day 14. During each of the subsequent ‘OFF’ periods there was an increase in sputum P aeruginosa bacterial counts towards baseline. In spite of the retention of increasing numbers of subjects who were no longer inhaling active therapy as the trial progressed, mean reductions in bacterial counts were still seen in each of the subsequent DRCFI treatment (‘ON’) periods.

Change in mean sputum Pseudomonas aeruginosa bacterial density across the 24 weeks of the study comparing DRCFI and placebo groups in the modified intention to treat (mITT) population. (Dotted line represents placebo, solid line represents DRCFI; note that data presented here are from both subjects who remained on trial drug and those who had withdrawn from trial drug due to pulmonary exacerbation; *p<0.05, **p<0.01, ***p<0.001 comparing DRCFI and placebo groups for change in bacterial density from baseline; CFU, colony forming unit; DRCFI, dual release ciprofloxacin for inhalation.)
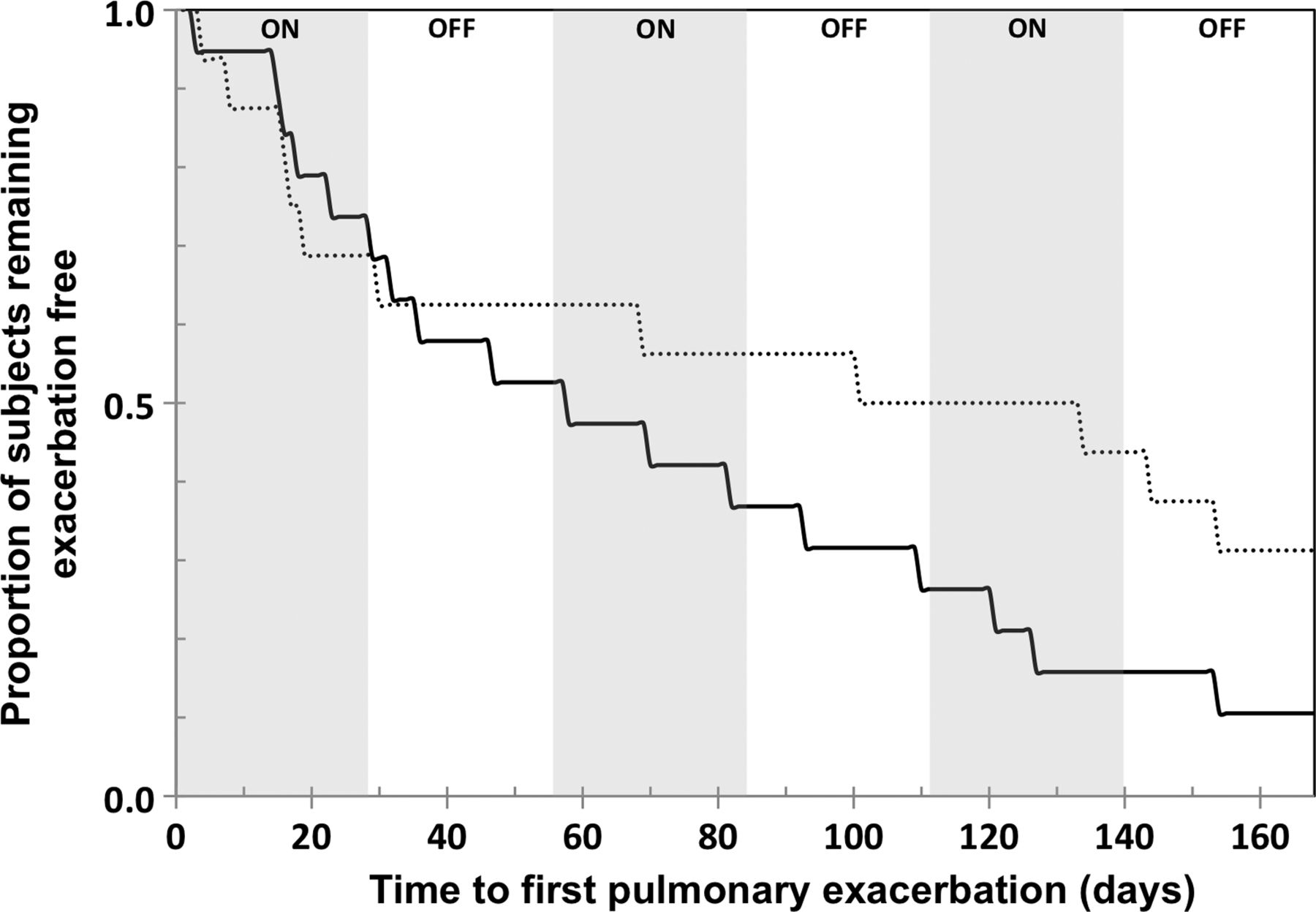
Pulmonary exacerbations were experienced by 17 (77%; all required antibiotic therapy) of placebo subjects and 11 (55%; 8 required antibiotic therapy) in the DRCFI group by day 168. Post hoc analysis revealed that the overall proportion of subjects requiring antibiotics for pulmonary exacerbation was lower in the DRCFI group (8 (40%) vs 17 (77%), OR 0.2, 95% CI 0.04 to 0.89, p=0.027 by Fisher's exact test). Intravenous antibiotics were received by three subjects in each group. The median time to pulmonary exacerbation by Kaplan–Meier analysis was 134 days for the DRCFI group and 58 days for the placebo group, although this achieved conventional statistical significance only on the per protocol population (p=0.057 mITT, p=0.046 per protocol, by log-rank test; see figure 3).

{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier curves comparing DRCFI and placebo groups for time to first pulmonary exacerbation in the modified intention to treat (mITT) population. (Dotted line represents DRCFI, solid line represents placebo; median 134 vs 58 days, p=0.057 mITT, p=0.046 per protocol, by log-rank test; DRCFI, dual release ciprofloxacin for inhalation.)
No significant differences were seen between the two treatment groups for changes to day 28 for other outcome measures including FEV1 (DRCFI −0.05±0.12 vs placebo 0.00±0.10 L, p=0.18), SGRQ total score (DRCFI −1.3±7.16 vs placebo −6.4±9.8, p=0.08) or 6MWT distance (DRCFI 0.6±71.6 vs placebo −7.6±92.3 m, p=0.54).
Failure to culture P aeruginosa at day 28 was seen more frequently in the DRCFI than placebo arms (12 (60%) vs 3 (14%), OR 9.5, 95% CI 1.8 to 63.0, p=0.003 by Fisher's exact test). New sputum pathogens were cultured at any time during the study in 12 placebo subjects (55%) on 21 occasions, the most common organisms being Stenotrophomonas maltophilia (four subjects), Pseudomonas fluorescens (three) and Streptococcus pneumoniae (two). New sputum pathogens were cultured in 9 (45%) DRCFI subjects on 16 occasions, the most common organisms being S maltophilia (five), Staphylococcus aureus (three) and Moraxella catarrhalis (two).
The identification of P aeruginosa isolates with lowered categorical susceptibility to ciprofloxacin (according to CLSI breakpoints) at any time during the study occurred in eight placebo subjects (38%, seven intermediate susceptibility and one resistant) and 10 DRCFI subjects (50%, six intermediate, four resistant). The change in P aeruginosa ciprofloxacin minimal inhibitory concentrations to day 28 (evaluating the most resistant isolate identified) did not differ significantly between the groups (median (range) 0 (−0.5 to +31) vs 0 (−0.75 to +0.5), p=0.26 by Mann–Whitney U test).
DRCFI was well tolerated with the overall incidence of AEs similar to the placebo group. Serious treatment emergent AEs were reported for three subjects in each arm (all events were pulmonary exacerbations)—none of the DRCFI events were considered treatment-related. Respiratory-related AEs leading to study discontinuation were more frequent in the placebo group (13 vs 7 patients) while non-respiratory AEs leading to discontinuation were similar (three for placebo: one anal ulcer, one sinusitis, one skin graft infection; four for DRCFI: two nausea, one sinusitis, one fatigue). Treatment emergent AEs occurring in more than three subjects in either arm were: lung disorder (DRCFI 11 subjects, 55% vs placebo 19, 86%), product taste abnormal (DRCFI four subjects, 20% vs placebo zero), nausea (DRCFI four subjects, 20% vs zero) and headache (DRCFI one subject, 5% vs placebo four subjects, 18%).
Discussion
In this phase II double-blind RCT, once daily, inhaled DRCFI demonstrated microbiological efficacy in non-CF bronchiectasis subjects with ciprofloxacin-sensitive P aeruginosa airway infection, without any evidence of tolerability or safety concerns. This microbiological efficacy, representing a greater than 10 000-fold fall in CFU (4 log fold drop), was associated with positive effects upon pulmonary exacerbation outcomes assessed by both time to first exacerbation (although statistically significant only in the per protocol population) and need for supplemental antibiotics. This is the first double-blind, placebo-controlled study of an inhaled agent to demonstrate clinical benefits in subjects with non-CF bronchiectasis, although it must be recognised that this was a specific, selected group of P aeruginosa-infected non-CF bronchiectasis subjects and hence the study results are not more broadly generalisable.
While the effects upon the primary outcome measure in ORBIT-2 were large and unambiguous, the effects upon pulmonary exacerbation outcomes need to be considered in the light of potential study limitations. First, the modest sample size creates the risk of Type 1 error. Second, the time to exacerbation endpoint was only conventionally statistically significant on the per protocol population (p=0.057 on mITT). Third, the DRCFI group were older and we cannot exclude that random imbalance in subject age at baseline may have influenced exacerbation outcomes in the two groups. However, baseline pulmonary exacerbation rates (a stratification variable at randomisation) were matched. Finally, the novel trial design employed in the current trial, in which subjects were withdrawn from study drug following first exacerbation, did reduce the potential to capture longer-term safety and tolerability data in some patients. However, this design safeguarded patients (given that many prior studies of inhaled therapies in this subject group have shown increased respiratory AEs)5–9 and yet ensured that the primary and key secondary (time to first pulmonary exacerbation) outcome measures were robustly captured. Furthermore, the potential duration of drug exposure in the current study was actually substantially longer than has been the norm in recent studies of inhaled antibiotics in both CF and non-CF bronchiectasis, which have mostly consisted of single 28-day antibiotic cycles.6 ,17–21
The antimicrobial efficacy of DRCFI developed early following commencement of therapy and was seen with each of the subsequent repeat treatment cycles. The reduction in bacterial density with DRCFI was similar in magnitude to that previously reported for TOBI in bronchiectasis subjects6 and substantially greater than the 2.2 log10/g reduction seen with TOBI in CF subjects after 4 weeks of therapy.4 However, in contrast to the prior study of nebulised TOBI in bronchiectasis,6 in ORBIT-2 there were fewer respiratory-related AEs and reductions in exacerbations in subjects receiving DRCFI compared with placebo. The recently reported single-blind study of low-dose inhaled gentamicin10 and double-blind RCT of inhaled ciprofloxacin dry-powder inhaler17 have also both demonstrated reductions in bacterial density of a similar magnitude to that reported here. However, both of those studies assessed this outcome in a range of airway pathogens in contrast to ORBIT-2 which assessed P aeruginosa alone.
DRCFI was well tolerated, without bronchodilator premedication. It is unclear whether this simply reflects improved tolerability of ciprofloxacin generally compared with aminoglycosides, given that ciprofloxacin dry-powder inhaler was not associated with bronchospasm in a recent study,17 or whether liposomal encapsulation may further improve tolerability.
Although there was no significant positive effect on quality of life to accompany the improvements in exacerbation data and respiratory AEs, this was not surprising as the study was not powered for this endpoint. Interestingly, two larger, recently reported studies of macrolides have also failed to show significant between-arm improvements in SGRQ scores in spite of significant improvements in a number of important clinical outcomes.21 ,22
In the current study, only subjects demonstrating at least one ciprofloxacin-sensitive P aeruginosa strain were enrolled and whether a significant benefit of therapy would emerge in subjects without any sensitive strains is unclear. However, we would anticipate that subjects with only resistant P aeruginosa strains on in vitro susceptibility testing will also derive clinical benefit from this formulation as DRCFI achieves high and sustained airway concentrations of ciprofloxacin that are likely to overcome even the most resistant strains.13 Furthermore, in studies of TOBI in CF, nearly 30% of subjects had tobramycin-resistant P aeruginosa and these subjects demonstrated similar clinical benefits to those with fully susceptible strains.4 Future studies evaluating the clinical efficacy of DRCFI in those without ciprofloxacin-sensitive P aeruginosa are needed.
The potential for development of antimicrobial resistance is inherent to any antibiotic therapy, particularly when used as a chronic maintenance therapy. While the current study did not demonstrate any significant increased resistance of P aeruginosa isolates to ciprofloxacin, some degree of selection pressure will result from the long-term use of any inhaled antibiotic including ciprofloxacin. For individuals with non-CF bronchiectasis, the critical question is whether the long-term use of inhaled antibiotics (including DRCFI) will result in improvements in clinical outcomes that outweigh the risks related to reductions in antimicrobial susceptibility. Only adequately powered long-term studies will address this question, although the data in subjects with CF suggest that the benefits of long-term nebulised antibiotics far outweigh any negative outcomes resulting from resistance induction.4 ,23 Until there are data to further inform this dilemma, it would be prudent to limit consideration of inhaled antibiotic therapies in non-CF bronchiectasis to those subjects experiencing the greatest morbidity, specifically P aeruginosa-colonised individuals with frequent infective exacerbations (ie, the population studied in ORBIT-2).
One further potential limitation of a strategy of maintenance, cycled, inhaled ciprofloxacin for bronchiectasis relates to the lack of an alternative, orally active antipseudomonal antibiotic class. Put simply, if a patient with bronchiectasis develops an infective exacerbation while on inhaled ciprofloxacin, would there be any added benefit in treatment with an oral fluoroquinolone or will subjects have an increased risk of subsequently requiring intravenous (non-fluoroquinolone) antibiotics for rescue? The current study was not powered to assess this. Future studies will evaluate this by determining whether subjects inhaling DRCFI who exacerbate recover more slowly with oral antipseudomonal therapy or are more likely to require rescue with intravenous antibiotics.
In the current study, once-daily inhaled DRCFI was well tolerated and demonstrated potent antipseudomonal microbiological efficacy in adults with non-CF bronchiectasis and airway infection by ciprofloxacin-sensitive P aeruginosa. Furthermore, positive effects upon pulmonary exacerbation outcomes were seen for the first time in a double-blind RCT of inhaled antibiotics in this patient group, although this was statistically significant only per protocol and may have been influenced by baseline age imbalance in this modest-sized phase II study. Confirmatory results in a larger data set are now required, which will also inform upon the competing risk of antimicrobial resistance.
Acknowledgments
The ORBIT-2 study group (all of whom recruited subjects and performed study related procedures) included Conroy Wong, Middlemore Hospital, Auckland, New Zealand; Gregory King, Woolcock Institute of Medical Research, Sydney, Australia; Paul King, Monash Medical Centre, Melbourne, Australia; Rob Stirling, The Alfred Hospital, Melbourne, Australia; Peter Wark, Centre for Asthma and Respiratory Disease, University of Newcastle, Newcastle, Australia; John Gillies, formerly of P3 Research, Tauranga, New Zealand.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors IG, PB, DC, DJS, DB and ADS participated in study design; DJS, PJT, JK and HG recruited study subjects and conducted study procedures; PB, IG, DC and DJS analysed and interpreted the results; DJS drafted and wrote the paper; all authors reviewed and revised the paper. All authors approved the final version.
-
Funding This study was funded by Aradigm Corp.
-
Competing interests This study was sponsored by Aradigm Corporation, Hayward, USA. The sponsor reimbursed organisations of members of the study group for all study related procedures (including DJS, PJT, JK and HG). DB has received fees for serving on the medical advisory board of Aradigm Corporation. None of the other investigators (or their organisations) received any additional monies including commissions, honoraria, travel fees, salary or research funds from Aradigm Corp. DJS has received fees for serving on the medical advisory boards of Pharmaxis, Phebra, GlaxoSmithKline and Boehringer Ingelheim and has received travel sponsorship from Boehringer Ingelheim to attend a scientific meeting. DB has received fees for serving on the medical advisory board of Aradigm Corporation. ADS has received fees for serving on the advisory boards of Forest Labs, Novartis and Bayer; lecture fees from Forrest Labs, GlaxoSmithKline, Chiesi, Novartis, AstraZeneca and Bayer; and support from GlaxoSmithKline, Boehringer Ingelheim and Chiesi for travel to attend respiratory symposia. PJT has received consulting fees from Merck Sharp and Dohme in 2011. JK and HWG have no conflicts of interests to declare. PB was an employee of Aradigm at the time the study was designed, conducted and analysed. IG and DC are employees of Aradigm and shareholders in the company.
-
Ethics approval Multicentre study—approved by ethics committees at each site.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves