Article Text

Abstract
Rationale Despite the potentially important roles for infection in adult non-cystic fibrosis (CF) bronchiectasis disease progression, the bacterial species present in the lower airways of these patients is poorly characterised.
Objectives To provide a comprehensive cross-sectional analysis of bacterial content of lower airway samples from patients with non-CF bronchiectasis using culture-independent microbiology.
Methods Paired induced sputum and bronchoalveolar lavage samples, obtained from 41 adult patients with non-CF bronchiectasis, were analysed by 16S ribosomal RNA gene pyrosequencing. Assessment of species distribution and dispersal allowed ‘core’ and ‘satellite’ bacterial populations to be defined for this patient group. Microbiota characteristics correlated with clinical markers of disease.
Measurement and main results 140 bacterial species were identified, including those associated with respiratory tract infections and opportunistic infections more generally. A group of core species, consisting of species detected frequently and in high abundance, was defined. Core species included those currently associated with infection in bronchiectasis, such as Pseudomonas aeruginosa, Haemophilus influenzae and Streptococcus pneumoniae, and many species that would be unlikely to be reported through standard diagnostic surveillance. These included members of the genera Veillonella, Prevotella and Neisseria. The comparative contribution of core and satellite groups suggested a low level of random species acquisition. Bacterial diversity was significantly positively correlated with forced expiratory volume in 1 s (FEV1) and bacterial community composition similarity correlated significantly with FEV1, neutrophil count and Leicester cough score.
Conclusions Characteristics of the lower airways microbiota of adult patients with non-CF bronchiectasis correlate significantly with clinical markers of disease severity.
- Respiratory Infection
- Bronchiectasis
- Bacterial Infection
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does the lower airway microbiota in non-cystic fibrosis bronchiectasis reflect clinical measures of disease progression?
What is the bottom line?
-
There is a significant correlation between airway microbiota composition and the severity of airways disease, cough-specific symptoms and airway inflammation.
Why read on?
-
We describe a relationship between airway microbiota and clinical measures that could provide a novel biomarker of disease, and also potentially indicate causal interactions
Introduction
Non-cystic fibrosis (CF) bronchiectasis (hereafter referred to as bronchiectasis) is a chronic airway disease characterised by abnormal destruction and dilation of the large airways, bronchi and bronchioles.1 As a result of the associated dysfunction of mucociliary clearance, a vicious cycle is established involving persistent bacterial colonisation, chronic inflammation of the bronchial mucosa, and progressive tissue destruction.2 Bronchiectasis is associated with chronic and frequently purulent expectoration, multiple exacerbations and progressive dyspnoea that can become disabling.1–3 While bronchiectasis can result from a variety of recognised aetiologies, it is often considered idiopathic.
A number of bacterial species have been associated with lower airway infection in bronchiectasis through traditional diagnostic culture-based detection; in particular Pseudomonas aeruginosa, Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus and Moraxella catarrhalis,4 with certain species having been shown to correlate strongly with clinical markers of disease. For example, P aeruginosa, associated with accelerated lung function decline in CF, also has a detrimental effect on prognosis in non-CF bronchiectasis.5 ,6
Given the role of bacterial infection, in sustaining chronic disease and in the incidence of acute exacerbation, obtaining a better understanding of the relationship between airway bacterial species and bronchiectasis is important. As routinely deployed, culture-based diagnostic microbiology is well suited to the detection of certain respiratory pathogens. However, other bacterial species, particularly those that are refractory to growth under standard diagnostic conditions, may go unreported. The application of culture-independent techniques that detect and identify bacterial species based on nucleic acid sequence signatures has revealed that substantial differences may exist between species reported by culture, and the total pool of species present in lower airway samples.7 In particular, the introduction of next-generation sequencing approaches has led to detailed characterisation of the bacterial species associated with the lower airways in a number of respiratory contexts, including CF,8–14 and chronic obstructive pulmonary disease.15–17
Importantly, such detailed microbiological characterisation also allows the definition of the airway ‘microbiota’ for a particular condition. Here, microbiota refers to the entire pool of bacteria occupying a given body site. The characteristics of the microbiota may have clinical importance that is distinct from that of its composite species. For example, the diversity of the microbiota present in the lower airways of adult patients with CF has been shown to be correlated to lung function.9 Further, by assessing the distribution and dispersal of microbiota within the patient population, it is possible to categorise the species detected as either ‘core’ which are common across patients and typically abundant or ‘satellite’, which are rarer with the patient group and typically of low abundance when detected. Such meta-analytical approaches have been applied previously in the context of CF lung infections,9 ,12 allowing the identification of a subset of bacterial species characteristic of this condition to be identified.
We hypothesised that application of culture-independent analysis to samples from the lower airways of adults with bronchiectasis would reveal the presence of a diverse bacterial microbiota, and that characteristics of the microbiota detected would correlate with clinical measures of disease.
To test these hypotheses, 16S rRNA gene 454 pyrosequencing analysis was applied to DNA extracted from bronchoalveolar lavage (BAL) and induced sputum samples from 41 adults with bronchiectasis.
Materials and methods
The BLESS study was a 12-month double-blind, randomised, placebo- controlled study of low-dose erythromycin in patients with non-CF bronchiectasis with at least two infective pulmonary exacerbations in the preceding 12 months.18 At baseline, a subgroup of 41 patients who had no evidence of additional (non-bronchiectasis) chronic respiratory disease were compared with a group of healthy volunteers (full details of inclusion and exclusion criteria for the BLESS study, the current subgroup and the control group are provided in the online data supplement). Patients with bronchiectasis and control subjects were lifelong non-smokers (<2 pack-year history), aged 18–85 years with no history of asthma or atopy, recent respiratory tract infection (within 4 weeks) or conditions with the potential to impact the safe performance of bronchoscopy. Patients with bronchiectasis had no evidence of (non-bronchiectasis) acute or chronic respiratory disease and were not receiving systemic corticosteroids. All subjects were macrolide naïve and no patients in the study were receiving nebulised antibiotics.
Normal control subjects had no evidence of respiratory disease, no history of use of any bronchoactive medications and normal spirometry (forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC) and ratio of FEV1:FVC all lying within the normal predicted range, no significant acute response to inhaled bronchodilator (BD) and no evidence of small airways obstruction). Full details of inclusion and exclusion criteria for both groups are provided in the online data supplement. The study was approved by the Mater Human Research Ethics Committee, Brisbane, and all subjects provided written, informed consent. Patients’ details are summarised in table 1, with full details shown in online supplementary table S1, and diagnostic microbiology data shown in online supplementary table S2. Details of control subjects are shown in online supplementary table S3.
Summarised characteristics of patients with non-cystic fibrosis bronchiectasis
Lower airway sample collection
A full description of the methodology used in the collection of BAL samples is provided in the online data supplement. Sputum induction with 4.5% hypertonic saline was performed between 48 h and 5 days prior to bronchoscopy, using methods previously described.19 The first collected sample was stored at −80°C for subsequent molecular analysis, and the subsequent three samples (10, 15 and 20 min) were pooled and processed for determination of non-squamous cell counts (see online data supplement for full details). All BAL and sputum samples were frozen rapidly at −80°C and subsequently transported to King's College London on dry ice for processing.
DNA extraction
Nucleic acid extractions were carried out using a combination of physical disruption and the phenol/chloroform extraction method, described previously.12 Full details of the methodology used are provided in the online data supplement.
Pyrosequencing
Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) was performed as described previously.12 A full description of the methodology is provided in the online data supplement. The sequence data obtained have been submitted to the Science Research Associates database, hosted by the National Center for Biotechnology Information, under the study accession number SRA066194.
Statistical analysis
To avoid potential biases in comparisons of diversity between local communities due to varying number of sequences per sample,20 a randomised re-sampling method, using three indices of diversity (taxa richness (S*), Shannon–Wiener index (H′) and Simpson's complement index (1−D)) were used as described previously.12 To partition bacterial species detected across all samples into core and satellite species groups, the Poisson distribution test was carried out as described previously.9 Regression analysis, coefficients of determination (r2), residuals and significance (p) were calculated using Minitab software (V.14.20, Minitab, University Park, Pennsylvania, USA). Boxplots were generated using the XLSTAT programme (V.2011, Addinsoft, Paris, France). Meta-analysis, using Hedges’ d effect size measure, was performed as previously described.12
The Bray–Curtis quantitative index of community similarity and subsequent average linkage clustering of community profiles were performed using PAST (Paleontological Statistics programme, V.2.16), available from the University of Oslo website link (http://folk.uio.no/ohammer/past). Similarity of percentages (SIMPER) analysis was used to determine the contribution of each species to the observed similarity between samples. SIMPER analyses were performed as previously described27 using Community Analysis Package (V.4, Pisces Conservation Ltd, Lymington, UK). Mantel and partial Mantel tests were performed as described previously22 using the XLSTAT programme.
Results
Patients
Paired BAL and induced sputum samples were collected from 41 patients with bronchiectasis (see table 1 for demographics). Aetiologies were idiopathic (26, 63%), post infectious (10, 24%) and pink disease (4, 10%). Standard culture of sputum reported P aeruginosa in 11 subjects (27%), H influenzae in 12 (29%) and ‘normal respiratory flora’ in the remainder.
PCR amplification
PCR amplification was achieved for 38 of the 41 induced sputum sample DNA extracts and 15 of 41 corresponding BAL sample DNA extracts. Four out of 16 control group samples provided a positive bacterial signal by PCR. No amplification was achieved for extracts from saline controls.
16S rRNA gene pyrosequencing
An average of 2753 16S rRNA gene sequences were obtained per sample (SD±1326), with 140 distinct bacterial species detected (see online supplementary table S4). The majority of species detected were associated with respiratory tract infections, opportunistic infections more generally, or the commensal populations of the oral cavity, and included aerobic and anaerobic species (see online supplementary table S4). The species detected included those commonly associated with chronic infection in bronchiectasis: H influenzae was detected in 34 of the 38 induced sputum samples (89.5%), P aeruginosa in 25 (65.8%), S pneumoniae in 31 (81.6%), S aureus in 7 (18.4%) and M catarrhalis in 10 (26.3%). Culture-based detection of P aeruginosa and H influenzae was confirmed in all cases, with culture-independent sequence-based detection in a further 15 and 22 patients, respectively.
Pyrosequencing was also performed on the four healthy control samples for which a positive bacterial PCR signal was obtained (two BAL samples and two induced sputum samples). Given the small number of control samples for which sequence data were obtained, it was not possible to compare species distribution with that obtained for bronchiectasis samples. However, the bacterial taxa identified in control samples, and their relative abundance, are shown in online supplementary table S6. In keeping with the analysis of bronchiectasis samples, substantially more sequences were obtained from the sputum sample compared with the BAL. In both sputum samples, low relative levels of S pneumoniae/pseudopneumonia (0.15% and 1.36% of sequences, respectively), and H influenzae/parainfluenzae (0.59% and 3.87%, respectively) were detected. No other bronchiectasis-associated species were detected.
Identifying ‘core’ bacterial species
Poisson distribution tests were used to determine which species detected were non-randomly or randomly distributed across patients, based on their detection in induced sputum samples (see online supplementary figure S1). The process provided an objective basis for partitioning species into core and satellite species groups, respectively. The core group was composed of 86 species compared with 54 species in the satellite group (figure 1). The membership of the core and satellite groups is indicated for the species detected in online supplementary table S4. SIMPER analysis of bacterial community similarity between patients was used to identify those species that contributed most to the similarity observed. These species are listed, in decreasing order of contribution, in table 2. Again, these species (all core group members) represent a mixture of respiratory pathogens and species associated with the oral cavity, with the greatest contribution to the observed similarity between samples due to the presence of H influenzae.
Similarity of percentages analysis of bacterial community similarity between patients
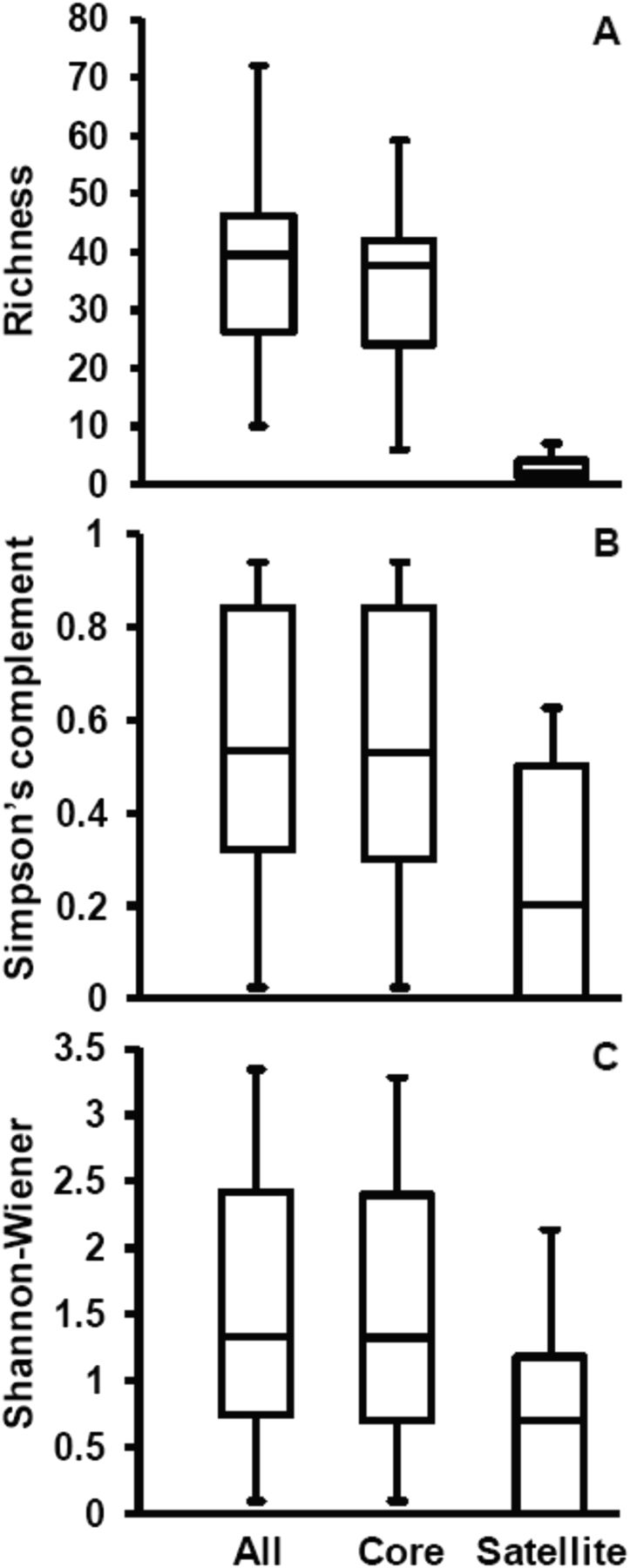
Boxplot comparisons of mean diversity in sputum samples for all communities and the core and satellite species groups across all patients (n=38). Three measures of diversity are given: (A) species richness, (B) Simpson's complement index (1–D), and (C) Shannon–Wiener index (H′). The top and bottom boundaries of each box indicate the 75th and 25th quartile values, respectively, and lines within each box represent the 50th quartile values. Ends of whiskers mark the lowest and highest diversity values in each instance.
Comparison of sample types
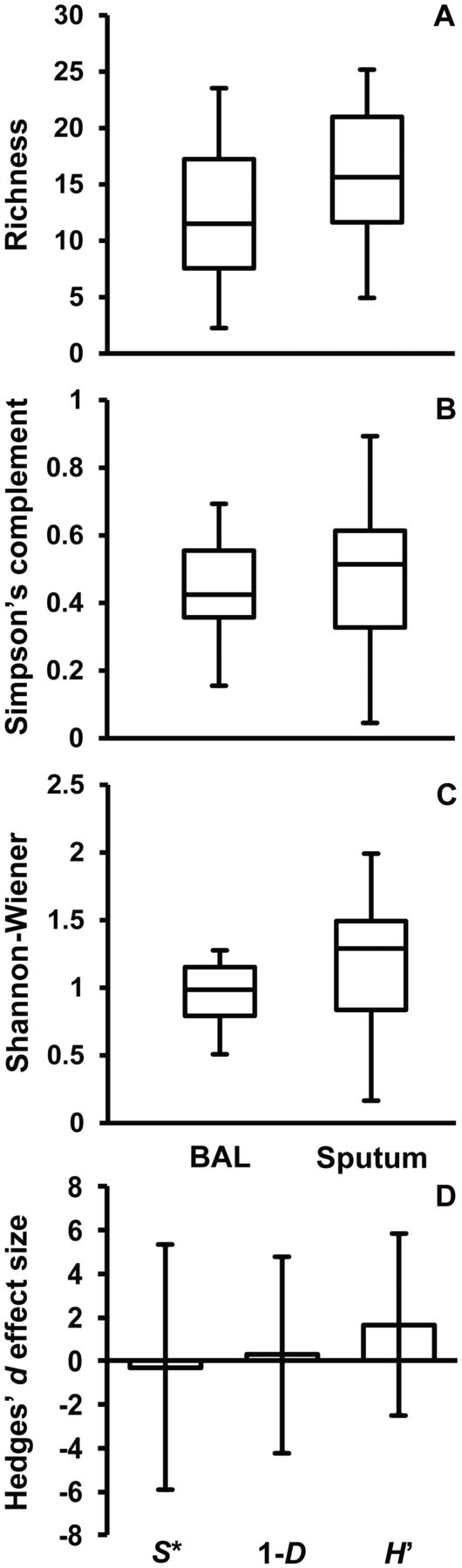
Analysis of bacterial diversity for induced sputum samples and BAL samples was performed to assess the degree to which the two sample types were comparable. Boxplot comparisons of mean diversity are shown in figure 2. Meta-analyses were used to summarise the effect of all 14 paired samples using Hedges’ d effect size measure,12 treating the different samples as independent of each other and the two sample types (BAL and sputum) as separate groups within samples. The overall effects of sample type on diversity showed no significant effect on any of the diversity measures used (figure 2). In addition, diversity was highly variable across individual samples, where meta-analyses of paired BAL and sputum samples revealed significant negative and positive effects on all diversity measures by sample type (see online supplementary figure S2).

Boxplot comparisons and meta-analysis of diversity between a subset of paired BAL and sputum samples (n = 14). Given are three measures of diversity; (A) species richness, (B) Simpson’s complement index (1-D), and Shannon-Wiener index (H). The top and bottom boundaries of each box indicate the 75th and 25th quartile values, respectively, and lines within each box represent the 50th quartile (media) values. Ends of whiskers mark the lowest and highest diversity values in each instance. (D) Overall meta-analysis for the three measures of diversity using Hedge’s d effect size measure between all BAL and sputum samples. Columns represent effect size and error bars represent the standard error (SE) of the effect size (n = 14). The SE bars that cross zero indicate no significant effect on diversity between samples types.
Correlation of bacterial community diversity and similarity with clinical measures
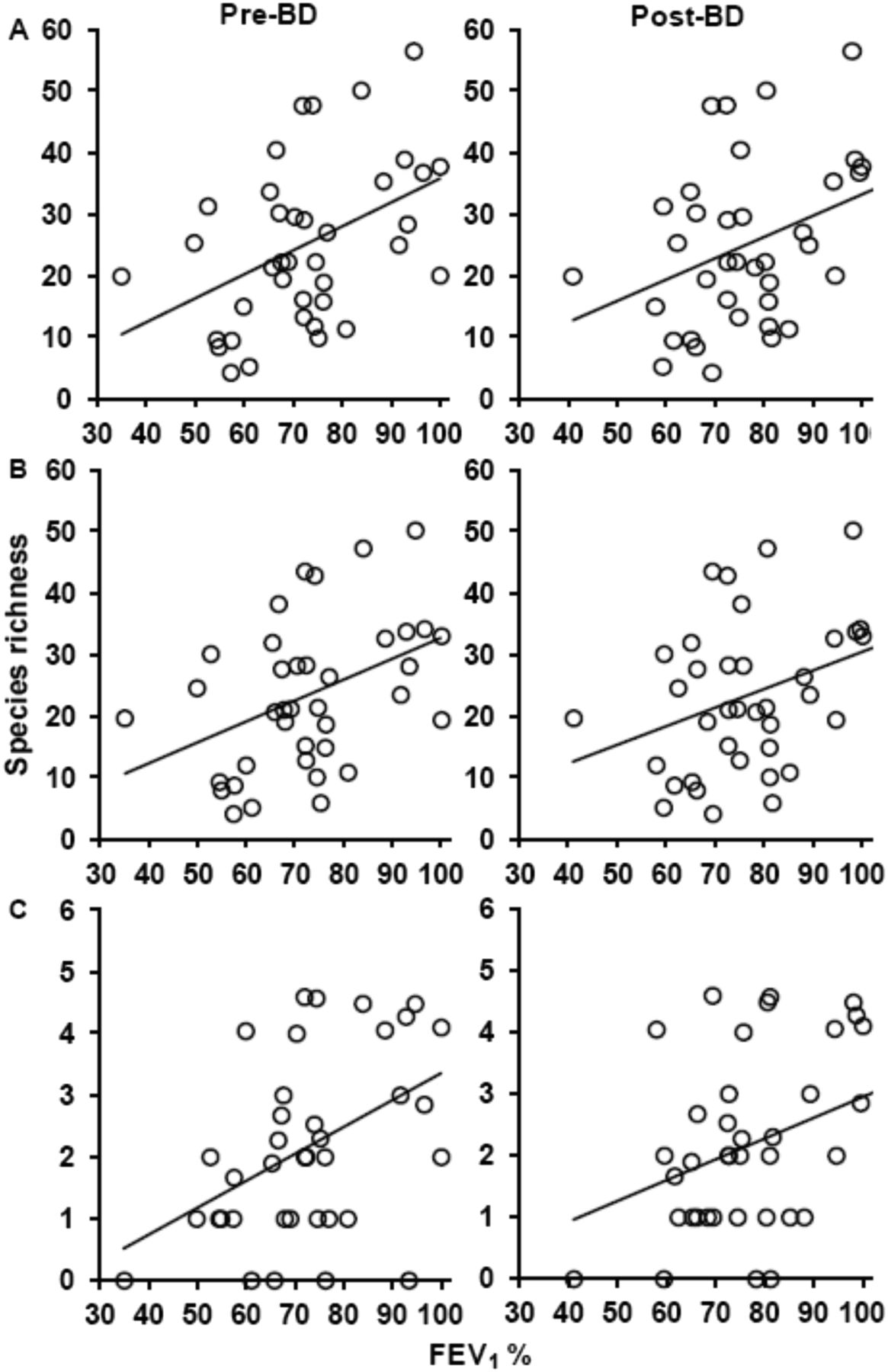
Bacterial community diversity was found to have a positive linear correlation with lung function (FEV1), as measured prior to, and after, administration of a BD (figure 3). This relationship held true regardless of whether analysis was performed on ‘all detected bacteria’, ‘core species’ or ‘satellite species’. Mantel tests were used to determine whether bacterial community composition similarity (Bray–Curtis similarity measure) correlated with differences in clinical measures of disease. A significant correlation (Mantel test) was observed between community composition similarity and FEV1 (pre and post BD) regardless of whether analysis was performed on ‘all detected bacteria’, ‘core species’ or ‘satellite species’. Significant correlations were also found between ‘all’ or core species with Leicester Cough Score (LCS), sputum neutrophil counts and gender, respectively. Mantel summary statistics are shown for all significant correlations in table 3. With the exception of gender, all other clinical measures were found to be co-correlated (see online supplementary table S5), reflecting their common relationship with airway disease. Partial Mantel tests revealed that FEV1, LCS and sputum neutrophil counts, and gender maintained significant relationships with bacterial community composition when controlling for the effects of each other clinical measure (see online supplementary table S6).
Mantel summary statistics for all communities, and the core and satellite species groups.
{kind=link}
{kind=link}
{kind=link}
The relationships between bacterial species richness and lung function (forced expiratory volume in 1 s (FEV1%)) for (A) all communities, (B) the core group and (C) the satellite species group. Relationships are given for pre-bronchodilator (BD) and post-BD values. In each case linear regression lines have been fitted. For pre BD: all communities, r2=0.20, F1,36=8.72, p=0.006; core group, r2=0.18, F1,36=7.69, p=0.009; and satellite group, r2=0.20, F1,36=8.87, p=0.005. For post BD: all communities, r2=0.14, F1,36=5.69, p=0.022; core group, r2=0.12, F1,36=5.09, p=0.030; and satellite group, r2=0.11, F1,36=4.20, p=0.048.
Discussion
The aims of this study were to characterise the bacterial microbiota associated with the lower airways of individuals with bronchiectasis, and to investigate whether a relationship exists between microbiota composition and clinical measures of disease. Before addressing these aims, it is important to consider the manner by which samples were obtained from the lower airways. Protected brush or BAL methods are often preferred due to the reduced potential acquisition of upper airway microbes. However, sputum induction is less invasive compared with bronchoscopy, and is more suitable for repeat sampling. The cross-sectional study presented here represents part of a larger, ongoing temporal study, and as such, an assessment of the relative suitability of the two sampling methods was performed. DNA extracts from induced samples were found to produce a positive PCR amplification signal significantly more frequently than extracts from corresponding BAL samples (93% compared with 37%, respectively). Comparison of the bacterial diversity reported in the respective extracts by pyrosequencing indicated that while the approaches differed in their sensitivity, their content was not significantly divergent. Given the greater coverage afforded by induced sputum, these samples were used for microbiota characterisation.
The pyrosequencing data presented here reveal a diverse airway microbiota, comparable to that reported in studies of other chronic respiratory diseases.8–17 Culture-based diagnostic microbiology is designed for the detection of specific pathogens of clinical significance, and as a result, the number of species reported is limited. The high frequency of detection of species traditionally associated with airway infections in patients with bronchiectasis reported here, including P aeruginosa, H influenzae, S pneumoniae, S aureus and M catarrhalis supports their perceived clinical importance. Further, the higher frequency of detection of P aeruginosa and H influenzae compared with culture-based diagnostic microbiology would appear to suggest that their prevalence, and their potential contribution to disease, may be yet greater than previously thought. In the case of H influenzae, some caution must be exercised when making comparisons between culture-based and molecular detection, as the reporting of the former was based on elevated levels compared with healthy individuals, rather than presence alone (as is the case for a number of other species, such as S aureus and certain streptococci). However, in the case of P aeruginosa, the magnitude of the difference was substantial, with its detection by molecular profiling alone in 14 of the patients studied (34%). The clinical implications of this are unclear, although they may be important given that our recent study suggests a particular subgroup benefit of erythromycin therapy in subjects with baseline P. aeruginosa infection (by standard culture based techniques).18 Furthermore, the detection by molecular methodologies of H influenzae in 83% of bronchiectasis patients in this study (compared with 29% by culture) is a novel finding whose clinical significance requires further evaluation.
Within the diverse array of bacterial species reported here, it was possible, based on distribution and dispersal, to categorise species as either core or satellite within the microbiota. The core species group identified here was substantially larger that the satellite species group, with the majority of species present well conserved across the patients sampled. The relative size of the core and satellite groups’ memberships also indicates a low level of random species acquisition, and potentially, a selective airway environment. The bacterial species that contributed most to the bacterial community similarity, as determined by SIMPER analysis, included a number of species that are considered to be respiratory pathogens and associated to some extent with bronchiectasis, including H influenzae, P aeruginosa and S pneumoniae. However, interestingly, many core species, making a substantial contribution to the similarity observed, have not previously been associated with bronchiectasis. For example, members of the genera Veillonella, Neisseria, Prevotella all made a substantial contribution to the core microbiota membership. In each case, these species would be unlikely to be reported through standard diagnostic microbiology as it is employed in the analysis of airway samples. Further, these are species that are commonly reported in culture-independent analysis of CF lower airway infections.9 ,10 ,23 ,24
The clinical significance of a much higher diversity of bacterial species present in the lower airways of patients with bronchiectasis than previously thought may extend beyond the pathogenicity of individual species. Bacterial diversity has been shown previously to be positively correlated with lung function in adult CF lung infections,9 where low diversity may reflect overgrowth by pathogens such as P aeruginosa. As with these previous studies, here we report a significant correlation between bacterial diversity and bacterial community similarity and FEV1.
By demonstrating that airway neutrophilic inflammation also relates to bacterial community similarity, our data extend the findings of prior studies that have shown relationships between airway inflammation and the bacterial load of potentially pathogenic species,25 and the presence of P aeruginosa in sputum.26 Hence the total bacterial community, not just specific presumed pathogenic bacteria, are related to neutrophilic airway inflammation in bronchiectasis.
Similarly, relationships recently described between the LCS and sputum bacterial load of pathogenic organisms26 can now be extended to bacterial community similarity. The strong relationship seen between this marker and LCS (but not daily sputum production or St George's Respiratory Questionnaire scores) raises the intriguing possibility that cough symptoms specifically are linked to particular airway bacterial communities.
The relationships observed between airway microbiota characteristics and measures of the severity of airways disease, cough-specific symptoms and airway inflammation mirror those reported in previous studies looking at airway disease in other contexts, such as CF, for which bacterial community composition has been found to be linked to clinical markers of disease progression.10 ,13 Should such relationships be found to exist consistently in patients with non-CF bronchiectasis, analysis of airway microbiota might be informative as to the extent of disease progression, and could therefore have the potential to serve as a disease biomarker.
This study has some limitations that should be considered. The description of the core airway microbiota presented here is derived from the set of samples that was analysed, and the extent to which it represents the wider population of patients with bronchiectasis is not established. For example, analysis was based on a subset of patients willing and able to undergo bronchoscopy safely and may differ somewhat from the broader population of patients with non-CF bronchiectasis. Further, the relatively mild average airways obstruction in this patient group should be taken into consideration when comparing the microbiota data with those derived from other patient populations. In fact, given the relationship between microbiota characteristics and markers of disease described here, we would expect that similar examinations of patients with bronchiectasis, stratified according to severity, or other disease facets, might report a range of core microbiota types.
This study focused on defining the core lower airway microbiota associated with bronchiectasis, and to examine the potential role of the microbiota as a pathogenic entity. However, this is not to detract from the potential contribution to airways disease of specific pathogens, such as non-tuberculous mycobacterial species or fungi. Assessing the relative contribution of such pathogens, and that of particular microbiota compositions, to disease progression in this context must now be investigated through longitudinal studies, especially those spanning clinically important events, such as exacerbations.
The manner in which the analysis was performed should also be considered. Here, bacterial detection was based on total extracted DNA. Other studies focusing on detection of airway bacteria have used propidium monoazide treatment of samples to limit the contribution of non-viable bacteria or extracellular DNA,12 or have based analysis on reverse transcribed rRNA.27 Such approaches can be particularly useful when there is substantial challenge to the bacterial populations being studied, for example, through antibiotic therapy. It was felt here though that a total DNA-based analytical approach, as an initial cross-sectional survey of airway microbiota, would provide the best opportunity for comparison with the growing literature, focusing on the study of bronchiectasis associated bacteria and the microbiota in lower airways disease more widely.
Here, we report a significant correlation between the composition of the lower airway microbiota in individuals with non-CF bronchiectasis and clinical measures of airways disease severity. The extent to which microbiota characteristics reflect various clinical aspects of disease, and the degree to which the core microbiota varies with patient demographics, now require further investigation. In addition, it is important to define the relationship between microbiota characteristics and clinically key events, such as the onset of periods of exacerbation and resulting antibiotic therapy, or infection by particular pathogens. The potential of the microbiota to play a causal role in disease progression also warrants investigation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All authors contributed equally to the conception, execution, analysis, and reporting of the study.
-
Funding CJvdG, LC and SKT are funded by the UK Natural Environment Research Council.
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.