Article Text

Abstract
Background Decreased activity of forkhead transcription factor class O (FoxO)3A, a negative regulator of NF-κB-mediated chemokine expression, is implicated in the pathogenesis of chronic obstructive pulmonary disease (COPD). Previously, we showed that quercetin reduces lung inflammation in a murine model of COPD. Here, we examined the mechanisms underlying decreased FoxO3A activation and its modulation by quercetin in COPD human airway epithelial cells and in a COPD mouse model.
Methods Primary COPD and normal human airway epithelial cells were treated with quercetin, LY294002 or erlotinib for 2 weeks. IL-8 was measured by ELISA. FoxO3A, Akt, and epidermal growth factor (EGF) receptor (EGFR) phosphorylation and nuclear FoxO3A levels were determined by Western blot analysis. Effects of quercetin on lung chemokine expression, nuclear FoxO3A levels and phosphorylation of EGFR and Akt were determined in COPD mouse model.
Results Compared with normal, COPD cells showed significantly increased IL-8, which negatively correlated with nuclear FoxO3A levels. COPD bronchial biopsies also showed reduced nuclear FoxO3A. Decreased FoxO3A in COPD cells was associated with increased phosphorylation of EGFR, Akt and FoxO3A and treatment with quercetin, LY294002 or erlotinib increased nuclear FoxO3A and decreased IL-8 and phosphorylation of Akt, EGFR and FoxO3A, Compared with control, elastase/LPS-exposed mice showed decreased nuclear FoxO3A, increased chemokines and phosphorylation of EGFR and Akt. Treatment with quercetin partially reversed these changes.
Conclusions In COPD airways, aberrant EGFR activity increases PI 3-kinase/Akt-mediated phosphorylation of FoxO3A, thereby decreasing nuclear FoxO3A and increasing chemokine expression. Quercetin restores nuclear FoxO3A and reduces chemokine expression partly by modulating EGFR/PI 3-kinase/Akt activity.
- Airway Epithelium
- COPD Mechanisms
- Oxidative Stress
- Cytokine Biology
- Complementary Medicine
- COPD Pathology
Statistics from Altmetric.com
- Airway Epithelium
- COPD Mechanisms
- Oxidative Stress
- Cytokine Biology
- Complementary Medicine
- COPD Pathology
Key messages
-
What are the mechanisms for reduced nuclear FoxO3A in COPD cells? Can the nuclear levels be modulated by neutraceutical Quercetin?
-
We found that aberrantly increased EGFR activity increases PI 3-kinase/Akt-mediated phosphorylation of FoxO3A, thereby decreasing nuclear FoxO3A and increasing chemokine expression. Quercetin restores nuclear FoxO3A and reduces chemokine expression partly by modulating EGFR/PI 3-kinase/Akt activity.
-
In this manuscript, we delineate the mechanisms by which FoxO3A activity is decreased in COPD cells and its contribution to chemokine responses. Importantly, we also show that quercetin modulates nuclear FoxO3A levels both in vitro and in vivo by decreasing EGFR/PI 3-kinase activities.
Introduction
Airways inflammation is one of the central features of chronic obstructive pulmonary disease (COPD). Airway epithelial cells (AEC) significantly contribute to inflammation by producing cytokines and chemokines in response to various stimuli, including cigarette smoke (CS). This in turn leads to recruitment of inflammatory cells to the airways. Under normal conditions, the expression of cytokines is tightly regulated by negative regulatory molecules, thereby limiting the influx of inflammatory cells and restoring the tissue homeostasis. Therefore, inactivation or reduced expression of negative regulatory molecules may result in sustained cytokine expression and development of airways inflammation. Previously, we demonstrated that AEC isolated from COPD patients express significantly more C-X-C chemokines than normal cells,1 suggesting that COPD cells are defective in regulating cytokine expression.
The forkhead transcription factor (Fox)O3A negatively regulates proinflammatory cytokine expression,2 in addition to playing a critical role in a wide variety of cellular processes, including immune regulation, and promoting resistance to oxidative stress.3 ,4 FoxO3A inhibits IL-8 expression in cells under oxidative stress by sequestering the RelA/p65 component of NF-κB, a transcription factor that stimulates IL-8 promoter activity.2 Recently, FoxO3A deficiency was shown to increase susceptibility to CS-induced lung inflammation in mice, but the mechanisms are not well understood.5
The regulation of FoxO3A activity is complex and primarily depends on post-translation modifications, such as phosphorylation, acetylation and ubiquitination, which affect its stability and subcellular localisation (nuclear vs cytoplasmic).6 Akt, a downstream target of PI-3 kinase, phosphorylates and promotes cytoplasmic accumulation of FoxO3A, ultimately leading to ubiquitination and degradation.7 FoxO3A that has been phosphorylated by Akt also binds to the 14-3-3 chaperone and remains in the cytoplasm.8 Phosphorylation of FoxO3A by Akt is also promoted by the acetylation of nuclear FoxO3A by CREB-binding protein combined with decreased SIRT1 (a type II histone deacetylase), which results in exclusion of FoxO3A from the nucleus.9 ,10 These observations suggest a critical role for Akt in regulation of FoxO3A nuclear localisation, but that role has not been established in AEC.
Increased Akt phosphorylation is seen in bronchial tissue of smokers11 and CS activates PI-3 kinase activity via EGFR activation.12 ,13 Phosphorylation of both EGFR and Akt is increased by oxidative stress, which has been implicated in the pathogenesis of COPD.14 ,15 Endotoxin, a cell wall component of Gram-negative bacteria that is present in appreciable amounts in CS,16 ,17 also increases Akt phosphorylation via EGFR activation.18 Based on these observations, we hypothesised that activation of the EGFR/PI 3-kinase/Akt signalling pathway decreases nuclear FoxO3A levels in COPD AEC, thereby increasing expression of proinflammatory cytokines, such as IL-8.
Quercetin (3,3′,4′,5,7-pentahydroxyflavone), a plant flavanoid, is a potent antioxidant. Quercetin also modulates activities of several lipid kinases, tyrosine kinases and serine/threonine kinases.19–22 Previously, we demonstrated that quercetin decreases chemokine levels and overall lung inflammation, and prevents progression of emphysema in mice displaying typical pathological and physiological features of COPD,23 ,24 but the mechanisms are not understood. The present study examines the mechanisms underlying the decreased nuclear FoxO3A levels in COPD AEC, and whether quercetin decreases chemokine expression by restoring nuclear FoxO3A levels. In this study, we also examined whether quercetin reduces chemokine expression in vivo by modulating nuclear FoxO3A levels.
Methods (details provided in online supplement)
Primary cells and bronchial sections
This study was approved by the University of Michigan Investigational Review Board. All bronchial segments were obtained from intact and healthy tissue of healthy non-smokers, smokers without COPD, or from COPD subjects during lung transplantation. Characteristics of COPD and normal donors are provided in online supplementary tables S1 and S2. AEC were isolated and cultured at air–liquid interface to promote differentiation into mucociliary phenotype as described.1 In some experiments, COPD mucociliary-differentiated AEC were treated with 1 µM erlotinib, LY294002 or quercetin for 2 weeks.
Bronchial tissue from healthy non-smokers, COPD or CF subjects obtained during lung transplantation was fixed in formalin and embedded in paraffin.
ELISA
Conditioned basolateral media, or lung homogenate supernatants, were used to quantify chemokine levels by ELISA.1 ,23
Transfection of 16HBE14o-cells
Immortalised 16HBE14o-AECs were reverse transfected with non-targeting (NT)- or FoxO3A-siRNA, and incubated for 2 days.25 Cell culture medium was used for IL-8 determination and cells for expression of FoxO3A protein.
Western blot analysis of nuclear, cytoplasmic and whole cell extracts
Nuclear and cytoplasmic protein extracts and total cell extracts were prepared as described previously.25 ,26 Aliquots of whole cell, cytoplasmic or nuclear extracts containing equal amounts of total proteins were subjected to Western blot analysis and specific bands quantified by densitometry.23 ,25
Immunodetection of FoxO3A and IL-8
Paraffin sections were deparaffinised, and subjected to immunofluorescence staining using antibody to FoxO3A as described previously,24 and/or IL-8 (R & D Systems, Minneapolis, Minnesota, USA), and sections were visualised under confocal microscopy (Carl Zeiss, Thornwood, New Jersey, USA).
Animals and treatment
Normal 8–10-week-old C57BL/6 mice (Charles River Laboratories, Wilmington, Massachusetts, USA) were exposed to elastase and LPS for four consecutive weeks and treated with quercetin or propylene glycol (vehicle), as described previously.23 ,24 Mice were exposed to cigarette smoke or room air for 6 weeks and treated with quercetin or vehicle during the last 2 weeks. Mice were then euthanased, and lung homogenates were used to prepare nuclear, cytoplasmic or whole cell extracts. All experiments described herein were approved by the Animal Care and Use Committee of the University of Michigan.
Statistical analysis
Results are expressed as means±SD. Data were analysed using SigmaStat statistical software (Systat Software, Inc, San Jose, California, USA). To compare two groups, an unpaired t test with Welch's correction was used. One-way analysis of variance (ANOVA) with Tukey–Kramer post-hoc analysis was performed to compare more than two groups. A ‘p’ value <0.05 was considered significant.
Results
Reduced nuclear FoxO3A correlates with elevated IL-8 expression in AEC
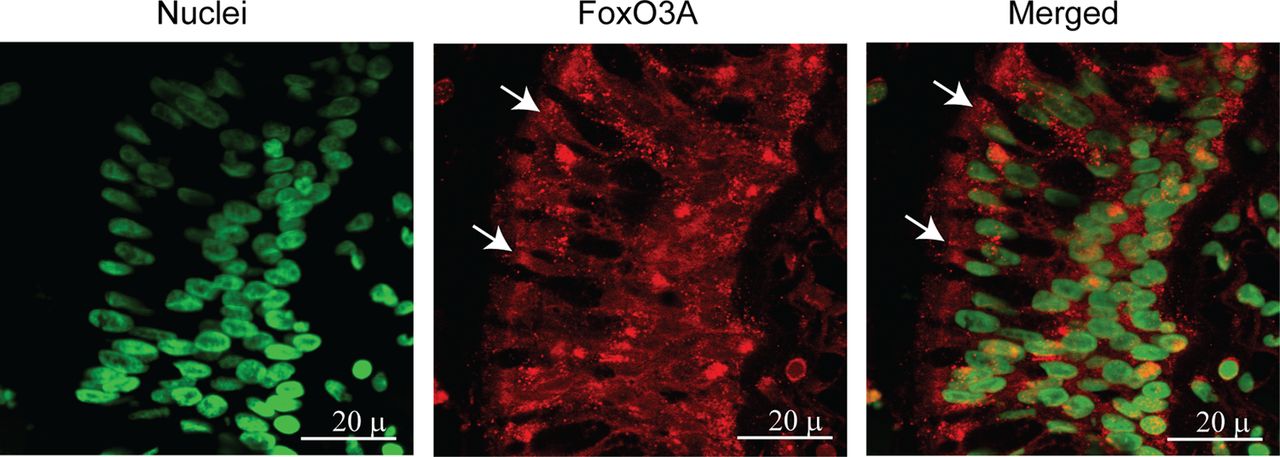
Both normal and COPD AEC grown at air–liquid interface showed mucocilliary differentiation (see online supplementary figure S1A) and transepithelial resistance of 400–600 Ω cm2. Compared with normal, COPD cultures also showed more periodic acid-Schiff stain (PAS)-positive cells (see online supplementary figure S1B). Immunofluorescence microscopy indicated that while normal cells show FoxO3A primarily in the nucleus, COPD AEC show majority of FoxO3A in the cytoplasm (figure 1A). Consistent with this, Western blot analysis showed significantly less nuclear FoxO3A in COPD AEC than in normal (figure 1B,C). By contrast, there was no difference in nuclear FoxO3A levels between AEC from healthy smokers and normal, therefore, subsequent experiments were performed with only normal and COPD cells. Despite reduced nuclear FoxO3A, COPD AEC showed total FoxO3A levels similar to normal AEC (figure 1D,E). In addition, COPD AEC showed increased IL-8 protein compared with normal AEC as observed previously1 (figure 2A). Importantly, nuclear FoxO3A levels showed a strong negative correlation with IL-8 protein between individual subjects (figure 2B). By immunofluorescence microscopy, IL-8 protein was observed in almost all cells in the cultures indicating that IL-8 expression is not specific to one cell type (figure 2C), however, COPD AEC showed comparatively more IL-8 protein than normal AEC.
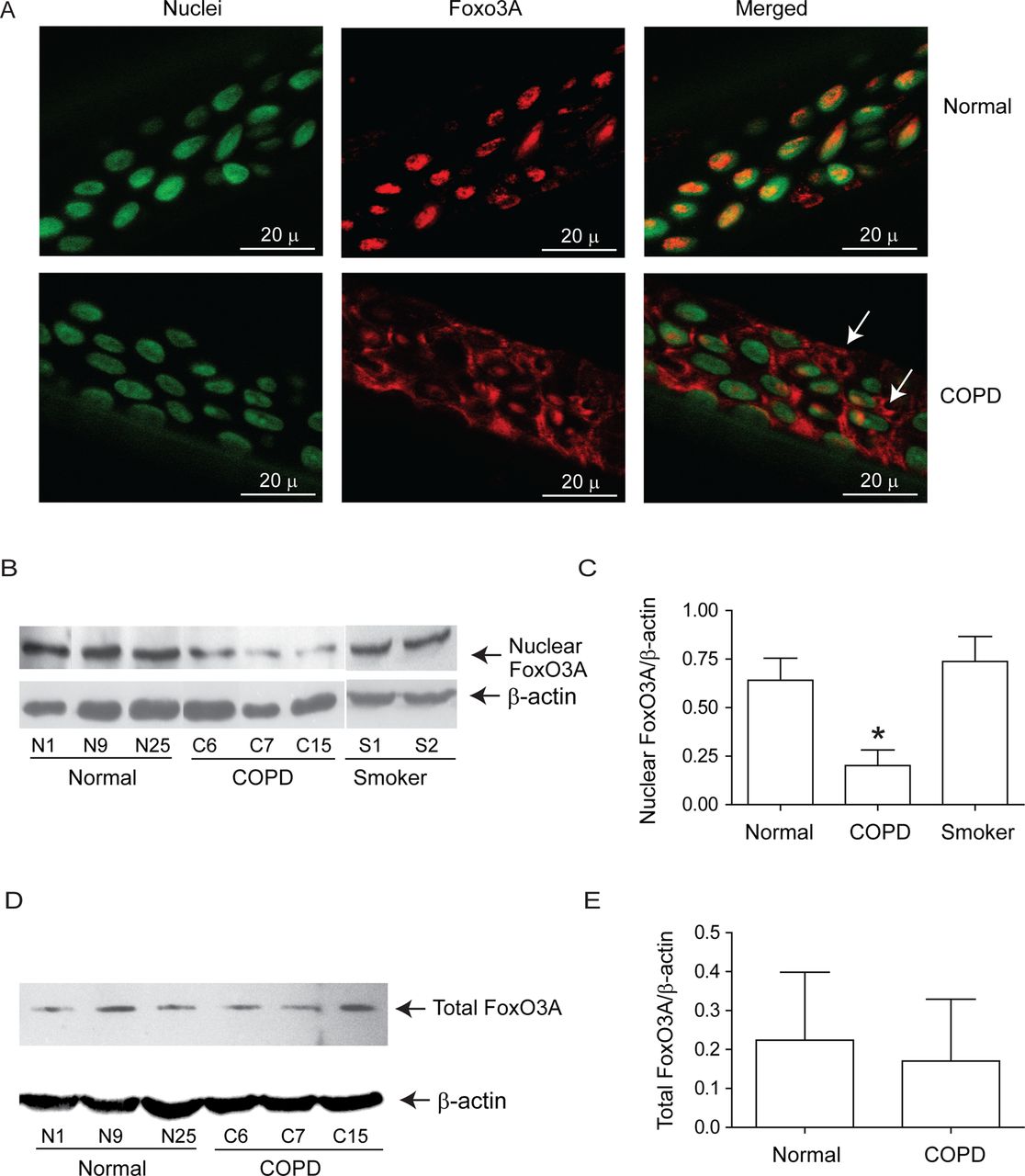
Nuclear FoxO3A is decreased in COPD AEC under basal conditions. Primary AEC from COPD or normal subjects were cultured in transwells at air–liquid interface to promote differentiation into mucociliary phenotype. (A) Cultures were fixed and embedded in paraffin. Five-micron-thick sections were deparaffinised, and incubated with antibody to FoxO3A. Bound antibody was detected by second antibody conjugated with Alexafluor-598 (red) and nuclei were counterstained with DAPI (green). Colocalisation of FoxO3A with nuclei appears yellow, and arrows point to cytoplasmic FoxO3A. Images are representative of AEC obtained from three COPD and three normal subjects. (B and C) Nuclear extracts from well-differentiated AEC cultures from healthy non-smokers, healthy smokers and patients with COPD were examined for FoxO3A by Western blot analysis, and band densities were quantified and expressed as fold increase over β-actin. (D and E) Total proteins from healthy non-smokers or COPD AEC were subjected to Western blot analysis with antibody to FoxO3A and levels of FoxO3A was normalised to β-actin. Data in C to E represents mean±SD calculated from cells isolated from three to six subjects per group; *, different from normal cells, p≤0.05, ANOVA.

IL-8 levels negatively correlates with nuclear FoxO3A. (A) Basolateral media from unstimulated COPD and normal AEC differentiated into mucociliary phenotype was used to determine IL-8 by ELISA. Data represents mean±SD calculated from cells isolated from six subjects per group; *, different from normal cells, p≤0.05; unpaired t test. (B) Correlation between nuclear FoxO3A (from figure 1C) and IL-8 was assessed by linear regression analysis (○ and • represent normal and COPD AEC, respectively). (C) Sections of COPD and normal AEC were probed with antibody to IL-8, bound antibody was detected by antimouse IgG labelled with Alexafluor-598 and sections were counterstained with DAPI to visualise nuclei. Arrows represents IL-8. Images are representative of three normal and three COPD cultures.
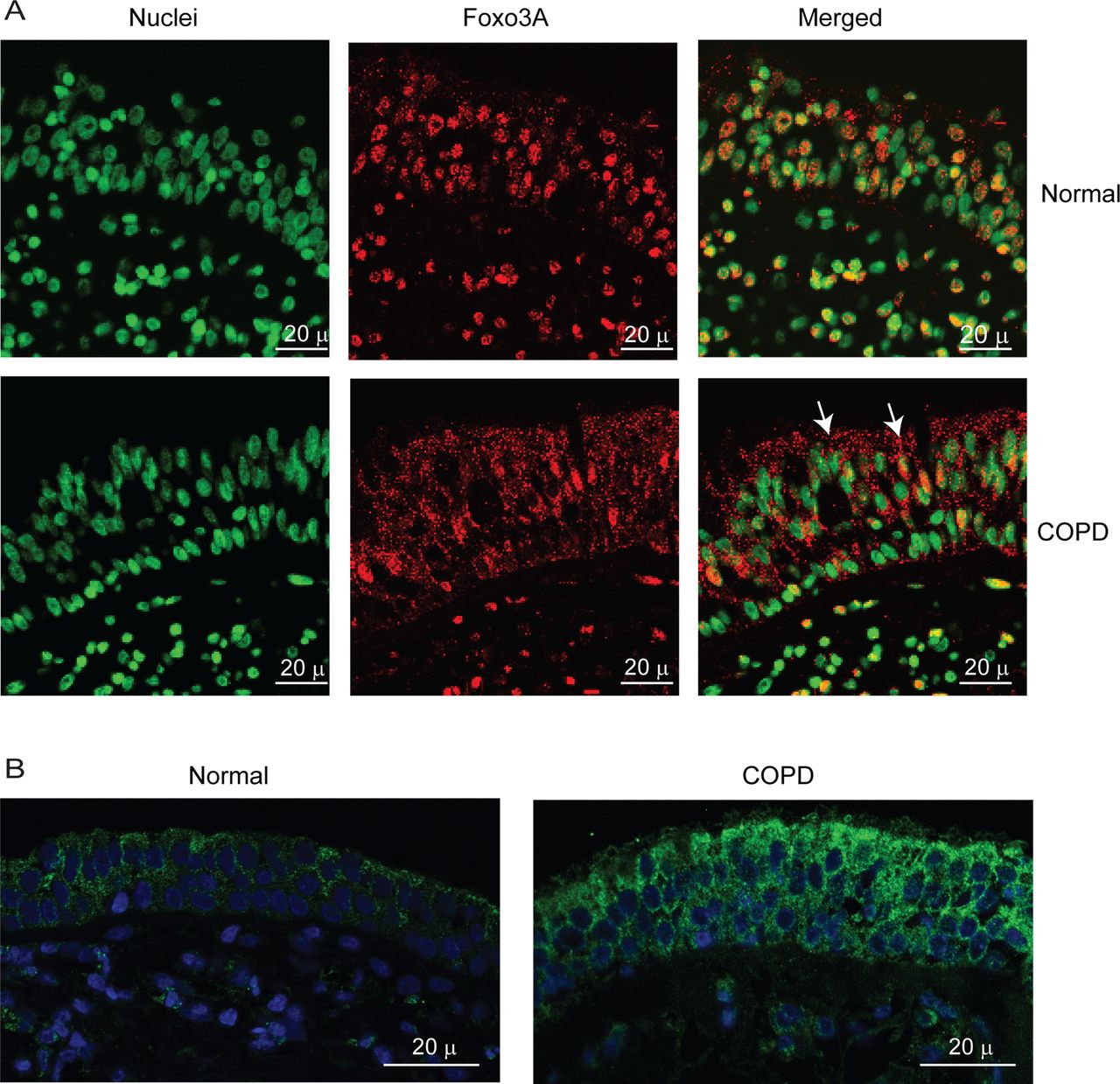
To exclude the possibility that the observed difference between normal and COPD AEC in nuclear FoxO3A was simply due to in vitro culturing conditions, we also immune-localised FoxO3A in bronchial sections obtained from COPD or normal subjects. In normal bronchial sections, the majority of FoxO3A was found to colocalise with the nucleus of bronchial epithelial cells (figure 3A), but in COPD sections, FoxO3A was mostly found in the cytoplasm and rarely in the nucleus. In both COPD and normal bronchial biopsies, IL-8 protein was observed in almost all the cells of bronchial epithelium, but the intensity was higher in COPD (figure 3B). These results indicate that cultured AEC truly represents the changes in FoxO3A distribution and IL-8 expression observed in vivo.

COPD bronchial epithelium shows decreased nuclear FoxO3A and increased IL-8 expression. Paraffin sections of bronchial tissue from COPD or normal subjects were deparaffinised, blocked and incubated with antibody to FoxO3A or IL-8. Bound antibodies were detected by antimouse or antirabbit IgG conjugated with Alexafluor-594, and nuclei were counterstained with DAPI. (A) FoxO3A and nuclei are represented by red and green, respectively; colocalisation of FoxO3A with nuclei appears yellow; arrows show FoxO3A in the cytoplasm. (B) IL-8 and nuclei are represented by green and blue, respectively. Images are representative of bronchial sections from three COPD and two normal subjects.
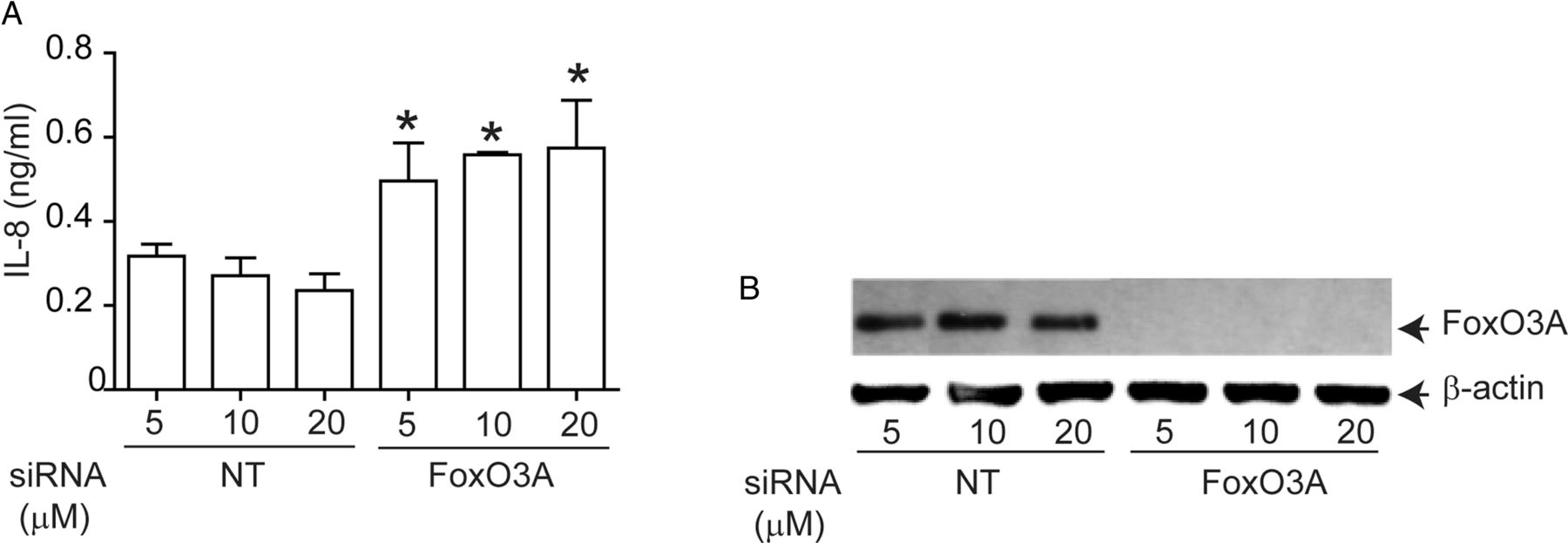
To determine whether FoxO3A regulates IL-8 expression in AEC, we knocked down FoxO3A using siRNA and measured basal IL-8 expression in immortalise AEC, because primary AEC are not readily amenable to transfection. Compared with NT siRNA-, FoxO3A siRNA-transfected cells showed significantly increased levels of IL-8 (figure 4A). FoxO3A siRNA- transfected, but not NT siRNA-transfected cells which showed complete knockdown of FoxO3A expression (figure 4B). These results confirmed that FoxO3A regulates basal IL-8 expression in human AEC.

Genetic inhibition of FoxO3A reduces IL-8 expression in normal airway epithelial cells. 16HBE14o-cells were reverse transfected with non-targeting (NT)- or FoxO3A-specific siRNA. Cells were incubated for 48 h, then media was changed and incubated for a further 24 h before culture supernatants were harvested and total cellular protein was isolated. (A) IL-8 levels measured by ELISA. Data represent mean and average from three independent experiments (*, different from cells transfected with NT siRNA, p≤0.05, unpaired t test). (B) FoxO3A protein levels by Western blot analysis. Representative of three independent experiments.
Akt phosphorylation is elevated in COPD AEC
Next, we examined whether reduced nuclear FoxO3A in unstimulated COPD AEC is due to increased phosphorylation. Cytoplasmic extracts from COPD AEC showed significantly increased phosphorylated FoxO3A compared with normal AEC (figure 5A,B). Because Akt, a downstream target of PI-3 kinase phosphorylates FoxO3A, we examined Akt phosphorylation and the effects of PI-3 kinase inhibitor, LY294002 on nuclear FoxO3A. Compared with normal, COPD AEC showed increased Akt phosphorylation (figure 5C,D) and treatment with LY294002-decreased phosphorylation of both Akt and FoxO3A, increased nuclear FoxO3A (figure 5E), and decreased IL-8 expression in COPD AEC (figure 5F). These observations imply that increased PI-3 Kinase/Akt activation is largely responsible for the observed nuclear FoxO3A reduction in COPD AEC.
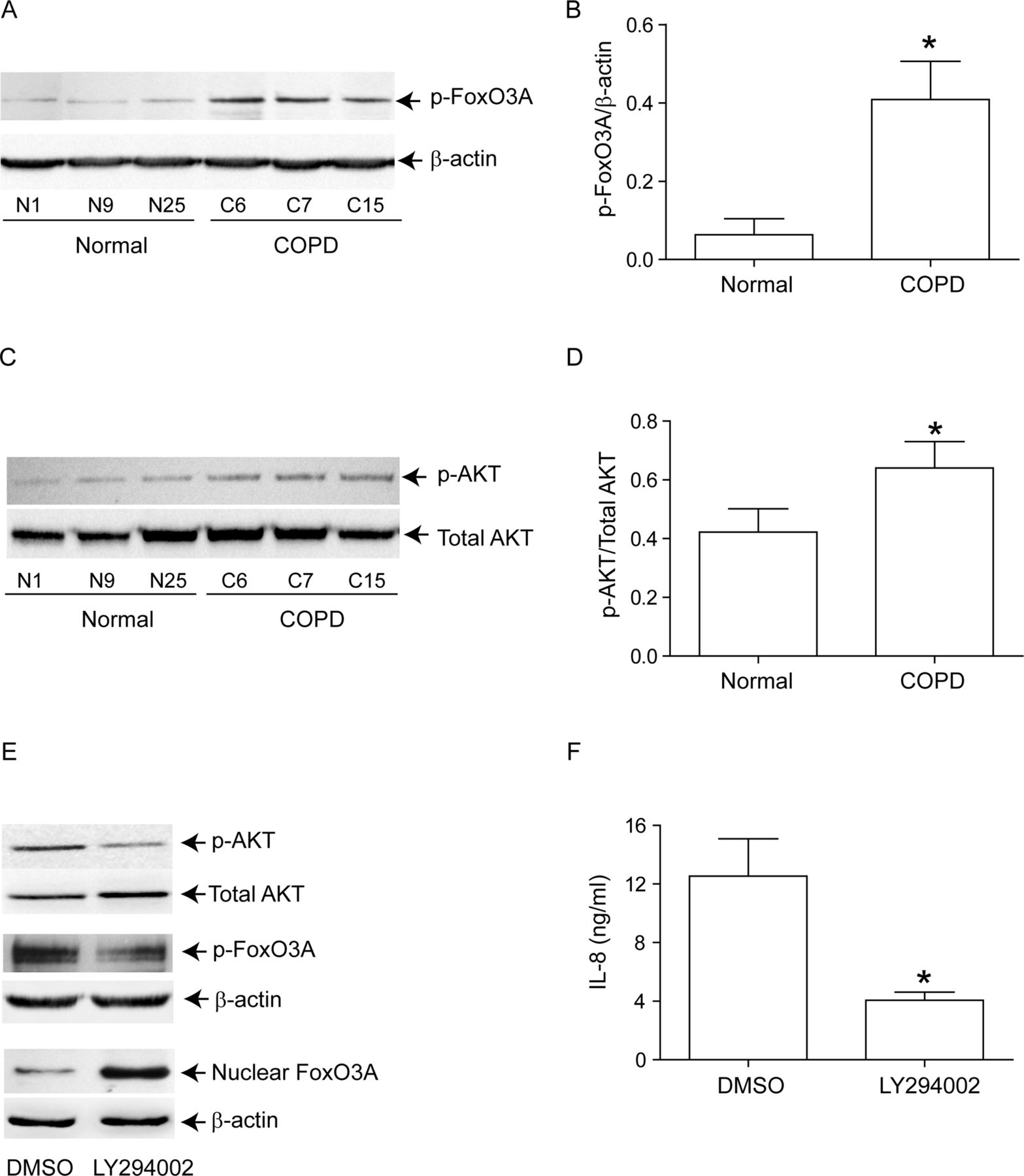
Inhibition of PI-3 kinase increases nuclear FoxO3A and decreased IL-8 in COPD AEC. Primary AEC from COPD subjects, or normal volunteers, were cultured as described in figure 1, then (A, B) cytoplasmic extracts were subjected to Western blot analysis with antibody to phospho-FoxO3A; (C, D) total cell lysates were subjected to Western blot analysis with antibodies to phospho-Akt, and total Akt and band densities were quantified by NIH image J. Data in B and D represents AEC from six subjects per group. (E) Mucociliary-differentiated COPD AEC were treated with DMSO (vehicle control) or 1 µM LY294002 for 2 weeks. Total proteins (for Akt), nuclear (for FoxO3A) or cytoplasmic (for phospho-FoxO3A) extracts were isolated from cells and subjected to Western blot analysis with relevant antibodies. Images are representative of three independent experiments. (F) IL-8 levels in the basolateral medium of the cells determined by ELISA; Mean±SD from three independent experiments, performed in duplicate with cells obtained from three donors (*, different from normal airway epithelial cells, p≤0.05, unpaired t test).
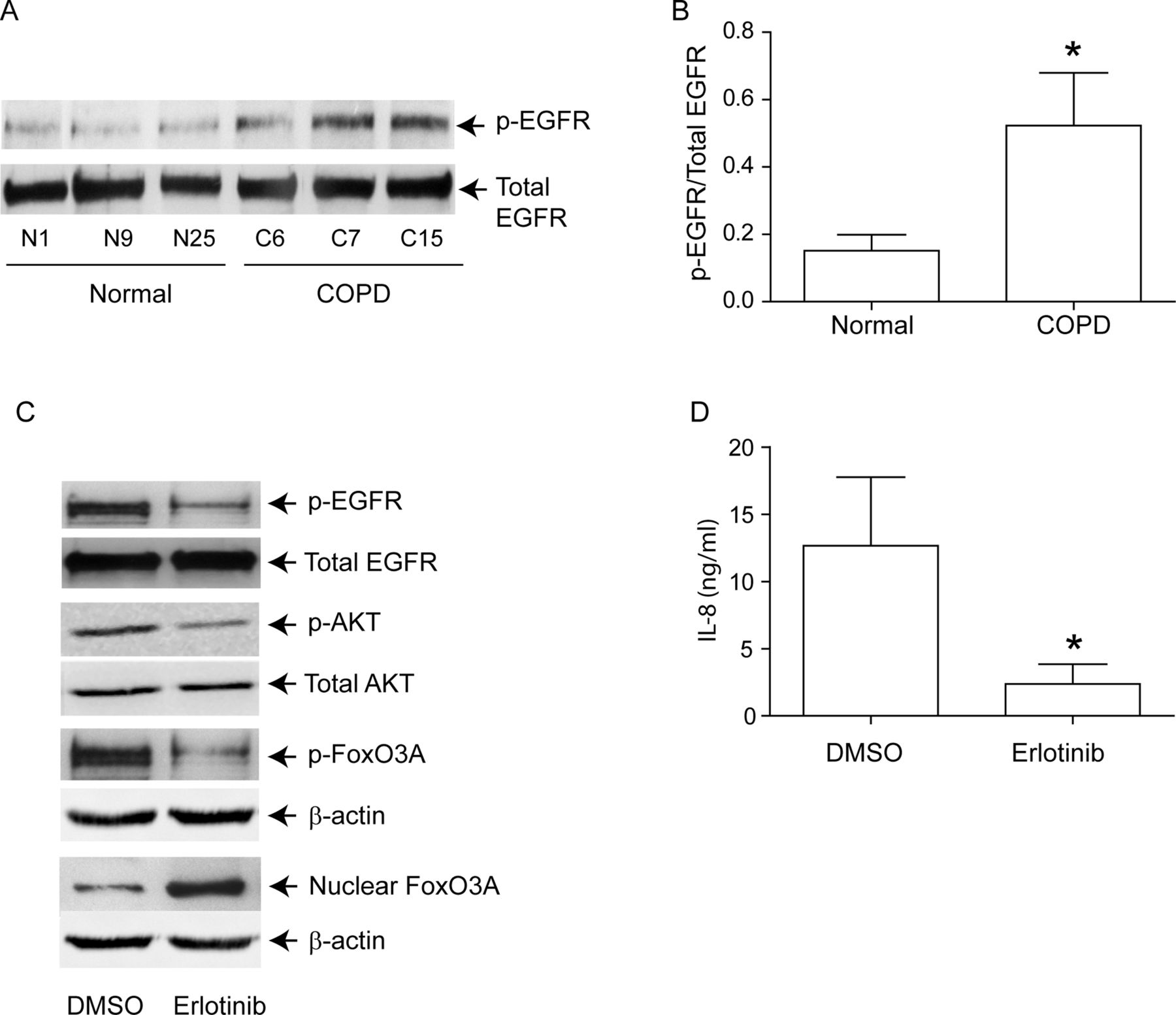
Increased EGFR activity correlates with Akt phosphorylation in COPD AEC
PI-3 kinase/Akt signalling pathway can be activated via tyrosine kinases, including growth factor receptors.27 Since EGFR activity is upregulated in COPD patients,28 and cigarette smoke increases EGFR activity,12 ,13 we next looked for evidence of elevated EGFR activity in COPD AEC. EGFR activity, as assessed by EGFR phosphorylation on tyrosine residue 1173, was higher in COPD than in normal AEC (figure 6A,B). Treatment with erlotinib, an inhibitor of EGFR decreased EGFR, Akt and FoxO3A phosphorylation, increased nuclear FoxO3A (figure 6C) and reduced IL-8 protein (figure 6D) in COPD AEC, implying that upregulated EGFR may be responsible for PI-3 kinase/Akt activation and subsequent decrease in nuclear FoxO3A, and increased basal IL-8 levels in COPD AEC.
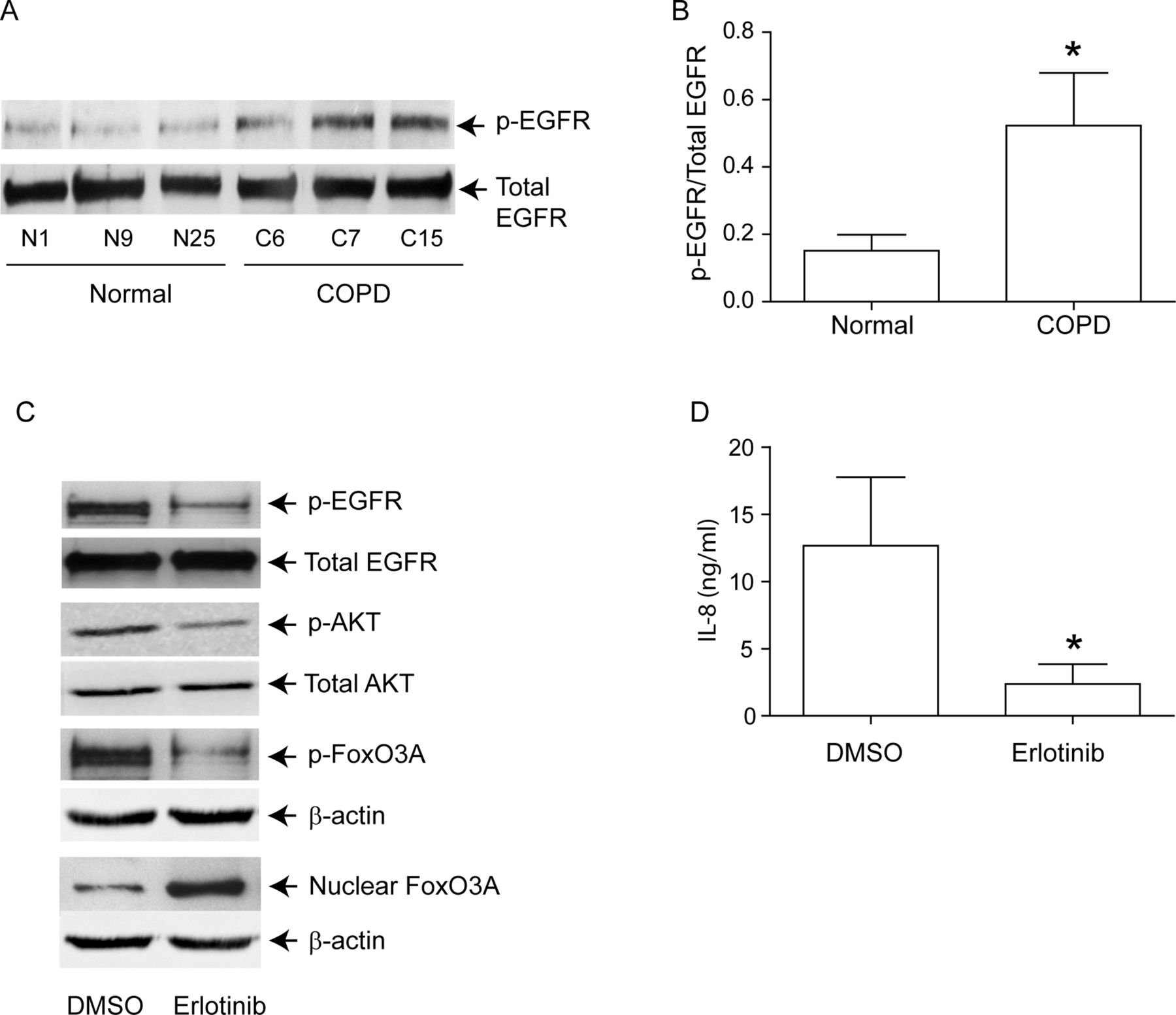
EGFR phosphorylation is increased in COPD cells, and inhibition of EGFR decreases IL-8 and increases nuclear FoxO3A in these cells. Primary human AEC from COPD subjects or normal volunteers were cultured as described in figure 1. (A, B) Total cell lysates were prepared and subjected to Western blot analysis using antibodies to phospho-EGFR and total EGFR. (A) Representative western blot; (B) phospho-EGFR/total EGFR ratios, n=6 per group. (C) mucociliary-differentiated COPD cells were treated with DMSO (vehicle control) or 1 µM erlotinib for 2 weeks. Total proteins (for EGFR), nuclear (for FoxO3A) or cytoplasmic (for phospho-FoxO3A) extracts were isolated from cells and subjected to Western blot analysis with relevant antibodies. Blots are representative of three independent experiments. (D) IL-8 levels in the basolateral medium were determined by ELISA. Data represents mean and SD calculated from three independent experiments performed in duplicate with cells obtained from three donors (* different from normal airway epithelial cells, p≤0.05, unpaired t test).
Quercetin treatment decreases IL-8 expression by inhibiting EGFR and Akt activation in COPD cells
One of the ways by which the anti-inflammatory nutriceutical quercetin appears to work is by blocking both tyrosine and threonine/serine protein kinases.19–22 We examined whether quercetin decreases IL-8 secretion in COPD AEC by reducing phosphorylation of EGFR, Akt and FoxO3A. Compared with DMSO-treated cells, COPD AEC treated with quercetin showed decreased IL-8 levels (figure 7A), reduced phosphorylation of EGFR and Akt, and increased nuclear FoxO3A (figure 7B). Increased nuclear FoxO3A in quercetin-treated COPD AEC was further confirmed by immunolocalisation (figure 7C). Thus, quercetin decreases IL-8 levels in COPD AEC is association with decreased activities of both EGFR and PI-3 kinase.

Quercetin treatment decreases phosphorylation of EGFR, Akt and FoxO3A and IL-8 levels in COPD airway epithelial cells. Primary AEC from COPD subjects were cultured as described in figure 1 and treated with DMSO (vehicle control) or 1 µM quercetin for 2 weeks. (A) IL-8 levels in the basolateral medium were determined by ELISA. Data represent mean and SD calculated from three independent experiments performed in duplicate or triplicate. (B) Total proteins (for EGFR and Akt), nuclear (for FoxO3A) or cytoplasmic (for phospho- FoxO3A) extracts were isolated from cells and subjected to Western blot analysis with relevant antibodies. Western blots are representative of three independent experiments conducted with cells obtained from three donors (* different from normal AEC, p≤0.05, unpaired t test). (C) Some cultures were fixed and embedded in paraffin. Five-micron-thick sections were used for immune-localisation of FoxO3A by confocal immunofluorescence microscopy. Colocalisation of FoxO3A with nucleus appears yellow, and arrows point to cytoplasmic FoxO3A. Images are representative of three independent experiments.
Quercetin treatment increases nuclear FoxO3A levels in elastase/LPS-exposed mice
To determine whether these findings were relevant in vivo, we examined our established murine model23 ,24 ,26 for changes in nuclear FoxO3A, and for the phosphorylation state of EGFR and Akt. Compared with PBS-exposed mice, elastase/LPS-exposed mice showed a significantly increased phosphorylation of both EGFR (figure 8A,B) and Akt (figure 8C,D) and reduced nuclear FoxO3A (figure 8E,F). Due to the non-availability of suitable antibody to mouse phospho-FoxO3A, we could not examine FoxO3A phosphorylation. Quercetin treatment significantly reduced phosphorylation of EGFR and Akt and increased nuclear FoxO3A in elastase/LPS-exposed mice (figure 9A), and also decreased the KC and MIP-2 to the levels that observe in PBS-exposed mice (figure 9B,C).

Elastase/LPS-exposed mice show decreased nuclear FoxO3A levels and increased phosphorylation of EGFR and Akt. Mice were exposed to elastase/LPS or PBS for 4 weeks. Lungs were excised, homogenised in PBS and aliquots of homogenates were used for either total protein (for EGFR and Akt) or nuclear protein (for FoxO3A) extraction. Aliquots equivalent to equal protein was subjected to Western blot analysis. Western blot images in A, C and E show samples analysed from three individual animals. Panels B, D and E represent ratios of p-EGFR/total EGFR, p-Akt/total Akt and FoxO3A/β-actin, respectively. Data represents mean and SD calculated from three independent experiments (n=6 mice * different from PBS-exposed animals, p≤0.05, unpaired t test).
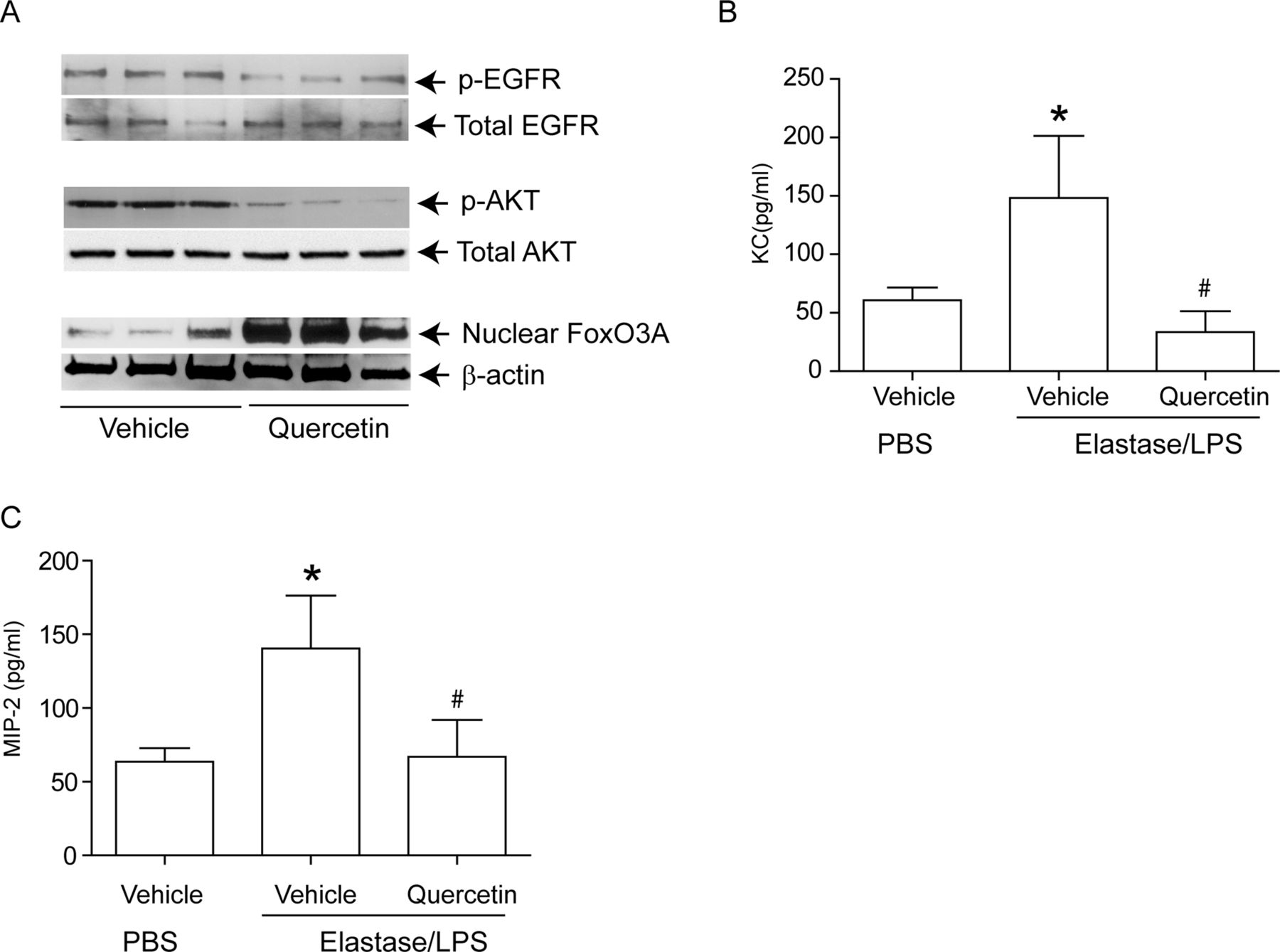
Quercetin treatment reduced chemokine expression in Elastase/LPS-exposed mice correlates with increased nuclear FoxO3A levels. Mice were exposed to elastase/LPS for 4 weeks and then treated with quercetin or vehicle for 2 weeks. PBS-exposed animals treated with vehicle were used as controls in some experiments. Lungs were excised, homogenised in PBS and aliquots of homogenates were used for either nuclear (for FoxO3A), or total protein (for EGFR and Akt) extraction. Aliquots equivalent to equal protein were subjected to Western blot analysis. Western blot images in A, B and C show samples analysed from three individual mice. (B, C) Lung homogenate supernatants were used for determination of chemokines by ELISA. Data represents mean and SD calculated from three independent experiments (n=6–9 * different from PBS-exposed animals treated with vehicle, p≤0.05; # different from elastase/LPS-exposed mice treated with vehicle p≤0.05, ANOVA with Tukey–Kramer post-hoc analysis).
We also examined the effect of quercetin in CS-induced changes in FoxO3A distribution in mice. Interestingly, CS-exposed mice showed nuclear FoxO3A, and KC and MIP-2 levels similar to room air-exposed mice (see online supplementary figure S2–D) and, not surprisingly, quercetin did not affect nuclear FoxO3A, KC or MIP-2 levels. By contrast, compared with room air-exposed mice, CS-exposed mice showed significant increase in MCP-1 levels and quercetin treatment reduced MCP-1 levels in CS-exposed mice (see online supplementary figure S2C), indicating that quercetin, as expected, negatively regulates NF-κB independent chemokine expression as well.
Nuclear FoxO3A levels are decreased in bronchial epithelium of CF patients
We determined the FoxO3A distribution in sections of bronchial biopsies from CF patients (who also show chronic lung inflammation) by immunolocalisation. Bronchial epithelium showed FoxO3A primarily in cytoplasm and rarely in the nucleus (figure 10), similar to that observed in COPD bronchial biopsies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Nuclear FoxO3A is decreased in CF bronchial epithelium. Paraffin section of bronchial tissue from CF patients was deparaffinised, blocked and incubated with antibody to FoxO3A. Bound antibody was detected by antirabbit IgG conjugated with Alexafluor-594, and nuclei were counterstained with DAPI. (A) FoxO3A and nuclei are represented by red and green respectively; colocalisation of FoxO3A with nuclei appears yellow; arrows show FoxO3A in the cytoplasm. Images are representative of bronchial sections from three CF patients.
Discussion
The principal findings of this study support a biochemical network in AECs of subjects with COPD by which persistent activation of the receptor, tyrosine kinase EGFR, mediates increased airway inflammation via activation of PI-3 kinase and Akt, consequent cytoplasmic retention of the anti-inflammatory transcription factor, FoxO3A, and increased basal levels of important neutrophil chemoattractant IL-8. Further, we show that low concentrations of quercetin, a non-toxic plant flavanol with antioxidant and kinase-inhibiting properties, block this pathway both in differentiated COPD AEC in vitro and in a murine model,23 ,24 ,26 that faithfully mimics many features of COPD pathology and physiology. These novel findings provide a mechanistic explanation for our previous observation of excessive C-X-C chemokine elaboration in COPD,1 and motivate additional study of a quercetin as a therapy to mitigate the development or progression of oxidative stress-induced lung damage.
These results complement and extend recent evidence for an important regulatory role for FoxO3A in COPD pathogenesis in humans,5 as it has in other chronic inflammatory diseases, such as inflammatory bowel disease and rheumatoid arthritis.29 ,30 FoxO3A-deficient mice develop spontaneous, multisystemic inflammatory syndrome, accompanied by NF-κB hyperactivity and increased proinflammatory cytokines.2 FoxO3A-deficient mice are also more susceptible to CS-induced lung inflammation and airspace enlargement.5 We extend the previous finding that Foxo3A expression is down-regulated in lung epithelial cells of COPD patients,5 by showing reduced nuclear FoxO3A localisation, which we confirmed in bronchial epithelium of COPD subjects in situ, and by demonstrating increased phosphorylated cytoplasmic FoxO3A in AEC from COPD patients. Interestingly, we found a similar decrease in nuclear FoxO3A in elastase/LPS-exposed mice, in which we have previously shown pathological changes typical of COPD including emphysema, extensive inflammation, remodelling of small airways, goblet cell hyperplasia, and susceptibility to viral and bacterial infections.23 ,24 ,26 By contrast, CS-exposed mice showed no changes in nuclear FoxO3A similar to that observed in AEC from healthy smokers, indicating the requirement for other endogenous/exogenous factors in addition to CS to reduce nuclear FoxO3A levels. These findings may partly explain why not all smokers develop COPD. Because FoxO3A interacts with RelA/p65 component of NF-κB in the nucleus, thereby inhibiting NF-κB binding to IL-8 promoter,5 it is not surprising that we observed increased IL-8 expression in COPD AEC. Increased IL-8 secretion has also been noted in CF airways, and the observation that nuclear FoxO3A is decreased in bronchial epithelium of these patients indicates that reduction in nuclear FoxO3A is not specific to COPD, but rather associated with chronic inflammatory conditions. Additional studies will be needed to define the possible consequences of altering nuclear FoxO3A levels on elaboration of other inflammatory mediators in the resting state, and in response to relevant proinflammatory stimuli or infection. Further clinical studies are required to establish the direct relationship of reduced nuclear FoxO3A or FoxO3A polymorphisms with disease severity in COPD patients.
Given the importance of FoxO3A shuttling between the nucleus and cytoplasm based on its phosphorylation state,10 identification of Akt and PI-3 kinase as mediators of FoxO3A regulation in AEC is important for a greater understanding of smoking or other environmental factors-induced lung disease. Akt phosphorylation is increased in the lungs and peripheral blood monocytes of COPD patients compared with normal subjects,31 and also increased by CS in both mice and in human AEC.11 ,32 Although CS can increase PI-3 kinase activity either by decreasing the expression of PTEN (phosphatase and tensin homologue deleted on chromosome 10),33 or via increasing the activation of EGFR, we believe that the latter may be a more relevant mechanism, because there was no decrease in the expression of PTEN in COPD AEC compared with normal (data not shown). By contrast, our finding of significantly increased EGFR phosphorylation with unchanged total EGFR levels in COPD cells compared with normal, and results using erlotinib, which inhibits EGFR specifically, both implicate EGFR activity for increased PI-3 kinase activity in COPD cells. Consistent with our observations, EGFR activity is increased in COPD patients,28 and CS stimulates PI-3 kinase/Akt activity via EGFR activation in vitro.12 ,13 Moreover, CS also increases LPS-stimulated EGFR activity in normal AEC.34 Although the epigenetic changes that maintain elevated EGFR activation in COPD AECs are not known at present, this study indicate the suitability of COPD AECs for understanding the biochemical mechanisms that mediate increased airway inflammation in COPD.
Quercetin has a broad spectrum of anti-inflammatory actions, dependent at least in part on its ability to block the ATP-binding sites of multiple lipid and protein kinases. Previously, we have demonstrated that quercetin decreases TNF-α-stimulated IL-8 production by blocking PI-3 kinase activity in human AEC,35 and significantly reduces chemokine expression and lung inflammation in elastase/LPS-exposed mice.23 ,24 We interpret the current data on the effect of quercetin on nuclear FoxO3A localisation as further support for a EGFR-PI-3 kinase-Akt pathway that ultimately regulates production of inflammatory mediators. However, one cannot rule out the possibility that quercetin may also increase nuclear FoxO3A levels by increasing SIRT1 levels, as we and others have shown that quercetin increases expression of SIRT1.23 ,36 SIRT1 deacetylates nuclear FoxO3A, particularly in cells undergoing oxidative stress, which show increased nuclear accumulation of protein acetylases, including CBP/ p300-associated factor.9 Deacetylation by SIRT1 prevents phosphorylation of FoxO3A by Akt and subsequent export of FoxO3A from nucleus to cytoplasm.
In conclusion, our studies provide an insight into one of the mechanisms by which IL-8 expression is upregulated in COPD airways in the stable state. We demonstrate that COPD AEC cultured in vitro retain their proinflammatory phenotype, with sustained increases in activity of EGFR and Akt, which in turn modulate nuclear FoxO3A levels. We show that a low dose of quercetin modulates the activities of EGFR and Akt, and inhibits chemokine expression both in vitro and in vivo. These results, together with our previous studies23 suggest that treatment with quercetin may reduce inflammation and improve lung function in COPD patients.
Acknowledgments
We acknowledge National Disease Research Interchange, Philadelphia, for providing tracheobronchial segments from healthy non-smokers and COPD subjects. We thank Marisa Linn for her assistance with processing tissues for immunofluorescence, and Drs M Hershenson and J Curtis for their valuable critiques while carrying out this work and also for critical reading of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Funding Supported by NIH grants AT004793, HL089772 (US).
-
Contributors SG, designed and performed the study and drafted manuscript; BLU assisted in performing cell culture experiments; ATC and KAA, provided technical support; PM, designed the study involving cigarette smoke-exposed mice, and provided facilities to expose mice to cigarette smoke; FJM, contributed intellectually and provided tissue samples; US, conceived and designed the study and finalised the manuscript.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval University of Michigan Investigational Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.