Article Text

Abstract
Background Current pharmacotherapy is highly effective in the clinical management of the majority of patients with stable asthma, however severe asthma remains inadequately treated. Prevention of airway remodelling is a major unmet clinical need in the management of patients with chronic severe asthma and other inflammatory lung diseases. Accumulating evidence convincingly demonstrates that activin A, a member of the transforming growth factor (TGF)-β superfamily, is a key driver of airway inflammation, but its role in chronic asthmatic airway remodelling is ill-defined. Follistatin, an endogenously produced protein, binds activin A with high affinity and inhibits its bioactivity. The aim of this study was to test the potential of follistatin as a therapeutic agent to inhibit airway remodelling in an experimental model of chronic allergic airway inflammation.
Methods BALB/c mice were systemically sensitised with ovalbumin (OVA), and challenged with OVA intranasally three times a week for 10 weeks. Follistatin was instilled intranasally during allergen challenge.
Results Chronic allergen challenge induced mucus hypersecretion and subepithelial collagen deposition which persisted after cessation of challenge. Intranasal follistatin (0.05, 0.5, 5 µg) inhibited the airway remodelling and dose-dependently decreased airway activin A and TGF-β1, and allergen-specific T helper 2 cytokine production in the lung-draining lymph nodes. Follistatin also impaired the loss of TGF-β1 and activin RIB immunostaining in airway epithelium which occurred following chronic allergen challenge.
Conclusions These data demonstrate that follistatin attenuates asthmatic airway remodelling. Our findings point to the potential of follistatin as a therapeutic for prevention of airway remodelling in asthma and other inflammatory lung diseases.
- Airway Epithelium
- Allergic lung disease
- Asthma
- Cytokine Biology
Statistics from Altmetric.com
Key messages
What is the key question?
-
To determine whether blocking activin A with its naturally occurring antagonist follistatin inhibits asthmatic airway remodelling.
What is the bottom line?
-
Follistatin instillation during allergen challenge inhibited secretion of activin A and transforming growth factor β1 in the lung, and significantly inhibited subepithelial collagen deposition and airway epithelial mucus production.
Why read on?
-
Our findings provide insight into the therapeutic potential of follistatin in the control of fibrosis in lung inflammatory disease, and highlight a role for activin A in the regulation of inflammation.
Introduction
Current pharmacotherapy is highly effective in the clinical management of the majority of patients with stable asthma, however severe asthma remains inadequately treated. Prevention of airway remodelling is a major unmet clinical need in the management of patients with chronic severe asthma and other inflammatory lung diseases.1 Increasing evidence implicates activin A, a homodimer of activin βA subunits and a member of the transforming growth factor (TGF)-β superfamily, in this process. Activin A can promote inflammation by stimulating production of inflammatory mediators including interleukin (IL)-1β, IL-6, tumour necrosis factor and nitric oxide. However, other studies show that activin A can inhibit inflammation.2 ,3 Clearly, regulation of the immunoregulatory effect of activin A is complex and dependent on the anatomical site, cell type and phase of the immune response. Importantly, activin A bioactivity is inhibited by the endogenously produced high-affinity binding protein follistatin.4
Accumulating data suggest that activin A regulates inflammation and fibrosis in the lung.5–9 In vitro, activin A stimulates proliferation of human lung fibroblasts and smooth muscle cells.5 ,10 In vivo, activin A is upregulated in bleomycin-induced pulmonary fibrosis, and follistatin injection inhibits this fibrosis.11 Furthermore, mice deficient in Smad3, common to the TGF-β/activin A signalling pathways, have decreased airway remodelling.12 In contrast, overexpression of the TGF-β/activin signalling intermediate Smad2 in airway epithelium induced activin A secretion and airway remodelling.13 Remodelling was inhibited by injection of activin A neutralising antibody prior to allergen challenge, providing direct evidence of a role for activin A in remodelling. However, detailed knowledge of the effect of inhibiting activin A with follistatin in chronic asthma is lacking. In the current study we investigated whether follistatin treatment during allergen challenge inhibited development of airway remodelling in a model of chronic allergic asthma. Our data identify activin A as an important driver of asthmatic airway pathology and highlight the therapeutic potential of follistatin as an inhibitor of airway remodelling.
Methods
Mice
Female BALB/c mice (7 weeks) were obtained from the Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria, Australia and housed in the Alfred Medical Research and Education Precinct (AMREP) animal house. All experimental protocols were approved by the AMREP Animal Ethics Committee.
Recombinant follistatin and follistatin luciferase bioassay
For follistatin production and assessment of bioactivity, see online supplement.
Immunisations and tissue processing
Mice were sensitised with intraperitoneal ovalbumin (OVA; A5503, Sigma-Aldrich, Saint Louis, Missouri, USA) in alum,6 and isofluorane-anaesthetised mice challenged intranasally (50 μl) with OVA (5 μg, 25 μg or 100 μg) three times per week.14–16 Groups were killed after 2, 5 or 10 weeks of allergen challenge, and 2 weeks after the final challenge (12 weeks) (see online supplementary figure S1A). For the follistatin instillation experiments, OVA-sensitised mice were challenged intranasally with OVA (25 μg) alone or mixed with follistatin (0.05, 0.5 or 5 μg), and analysis performed after 5 weeks of challenge (see online supplementary figure S1B). Controls received saline instead of OVA or follistatin. Bronchoalveolar lavage (BAL), differentials and tissue sampling were as described17 (see online supplement).
Cytokine ELISPOT, OVA-specific IgE, activin A, TGF-β1 and IL-13 ELISA
IL-4, IL-5 and IL-13 enzyme-linked immunosorbent spot (ELISPOT) on lung-draining lymph node (LN) cells were performed as described.6 OVA-specific IgE was detected as described.17 Activin A was measured by a specific ELISA as described.6 ,18 TGF-β1 was detected using a TGF-β1 ELISA kit (#DY1679, R&D Systems, Minneapolis, MN, USA) (see online supplement). IL-13 was detected using the IL-13 Ready-SET-Go! kit (#88-7137-88; eBioscience, Inc., San Diego, California, USA).
Follistatin radioimmunoassay
Follistatin concentrations were measured using a discontinuous radioimmunoassay as described.18
Activin A, TGF-β, follistatin and activin receptor immunohistochemistry
Immunohistochemistry was performed on 3 μm formalin-fixed sections. .Activin A and follistatin immunohistochemistry were performed as described.6 For TGF-β and activin receptor immunostaining antigen retrieval was performed and endogenous peroxidise blocked in H2O2. Sections were stained with antibodies specific for TGF-β1 (sc-146; Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA), or ActRIB/ALK4, ActRIIA and ActRIIB (N-20, sc-11984; N-17, sc-5667; and N-16, sc-5665, respectively; Santa Cruz Biotechnology, Inc.) or appropriate isotype controls. See online supplement.
Quantitative image analysis
The frequency of periodic acid Schiff (PAS), activin A, follistatin or ActRIB positive airway epithelial cells per mm basement membrane was calculated (http://fiji.sc). Alternatively, airway epithelial TGF-β, activin A, ActRIIA and ActRIIB immunoreactivity was expressed as integrated density (n=5–19 airways/mouse). The percentage of subepithelial collagen in Masson's trichrome-stained sections was calculated to a depth of 20 μm below the basement membrane.16 Smooth muscle thickness was measured at right angles across the muscle bundle on ActRIB-stained sections (n=2–7 airways/mouse). See online supplement.
Statistics
Data were analysed for normality and log transformed as necessary prior to analysis by analysis of variance and Tukey post tests or t tests (GraphPad Prism V.5.03). Differences were considered significant at p<0.05. Group sizes are indicated in the figure legends. All values in figures are mean±SEM.
Results
Repeated allergen challenge induces acute resolving pulmonary inflammation
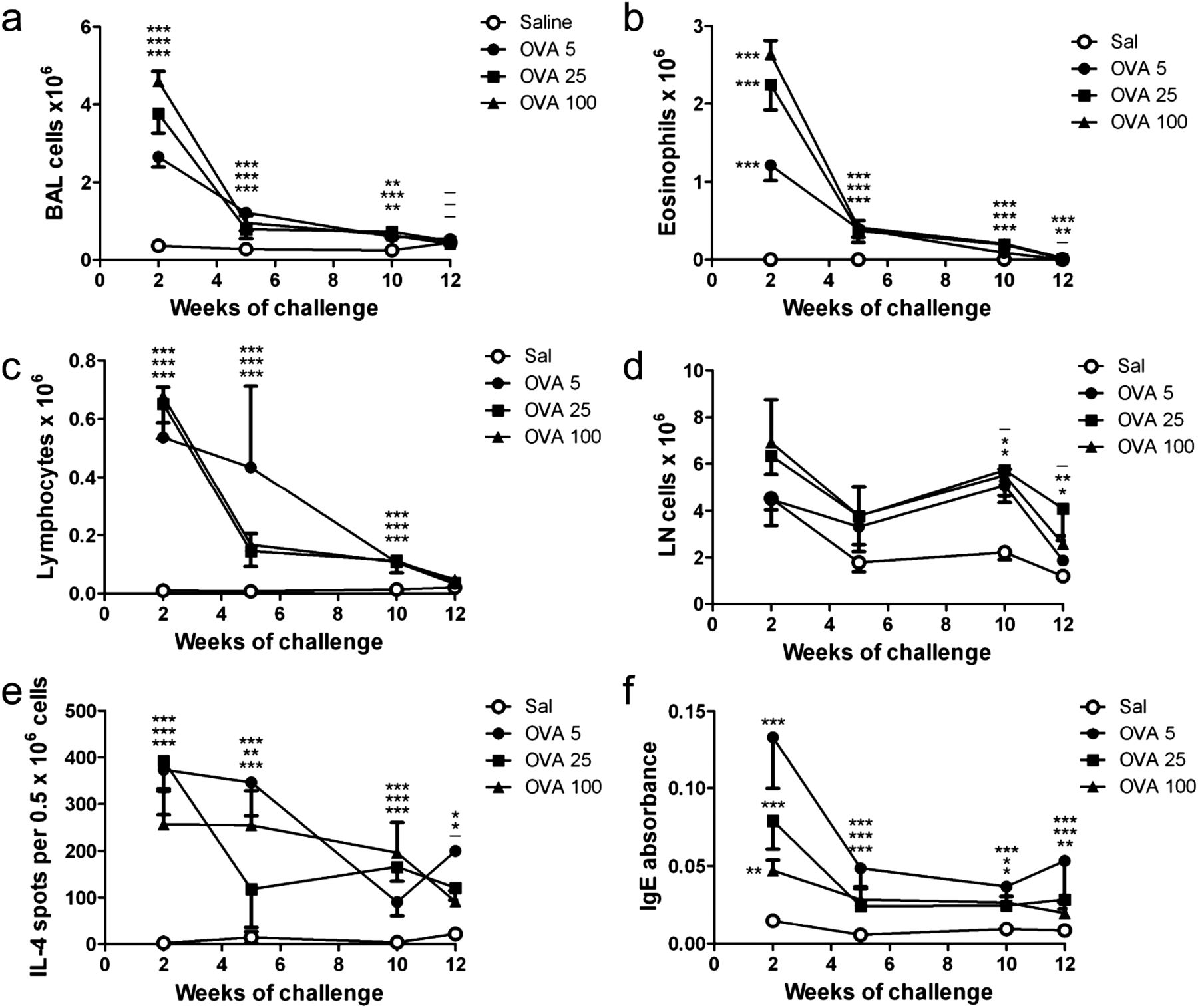
We investigated the effect of chronic allergen challenge on the kinetics of airway inflammation and remodelling to validate our model. Mice were challenged with 5, 25 or 100 μg OVA to investigate the effect of allergen dose (see online supplementary figure S1A). Regardless of OVA challenge dose, BAL cell counts were markedly increased compared with saline controls after 2 weeks of challenge, with eosinophils the major infiltrating cell type (figure 1A,B). After 5–10 weeks of challenge total BAL and eosinophil counts had decreased sharply but remained significantly higher than saline controls, and returned to baseline after challenges ceased. BAL lymphocytes followed a similar pattern (figure 1C). Cell counts in the lung-draining LN were elevated from 5 weeks and trended toward control values once allergen challenges stopped (12 weeks) (figure 1D). The frequency of IL-4-producing cells in the draining LN peaked after 2 weeks of challenge, decreasing gradually thereafter, but remaining above saline levels at 12 weeks (figure 1E). OVA-specific IgE levels were significantly elevated after 2 weeks of challenge, dropping sharply by 5 weeks and remaining stable thereafter (figure 1F). Thus allergen challenge induced acute inflammation at 2 weeks, similar to other acute asthma models, but this decreased sharply upon continued OVA challenge, and decreased further once allergen challenge ceased. Airway inflammation/eosinophilia was dose dependent, with the 100 μg group showing the greatest inflammation. The dose-dependent effect on serum OVA-specific IgE was reversed, with the 5 μg OVA challenge group having the highest level at all time points.
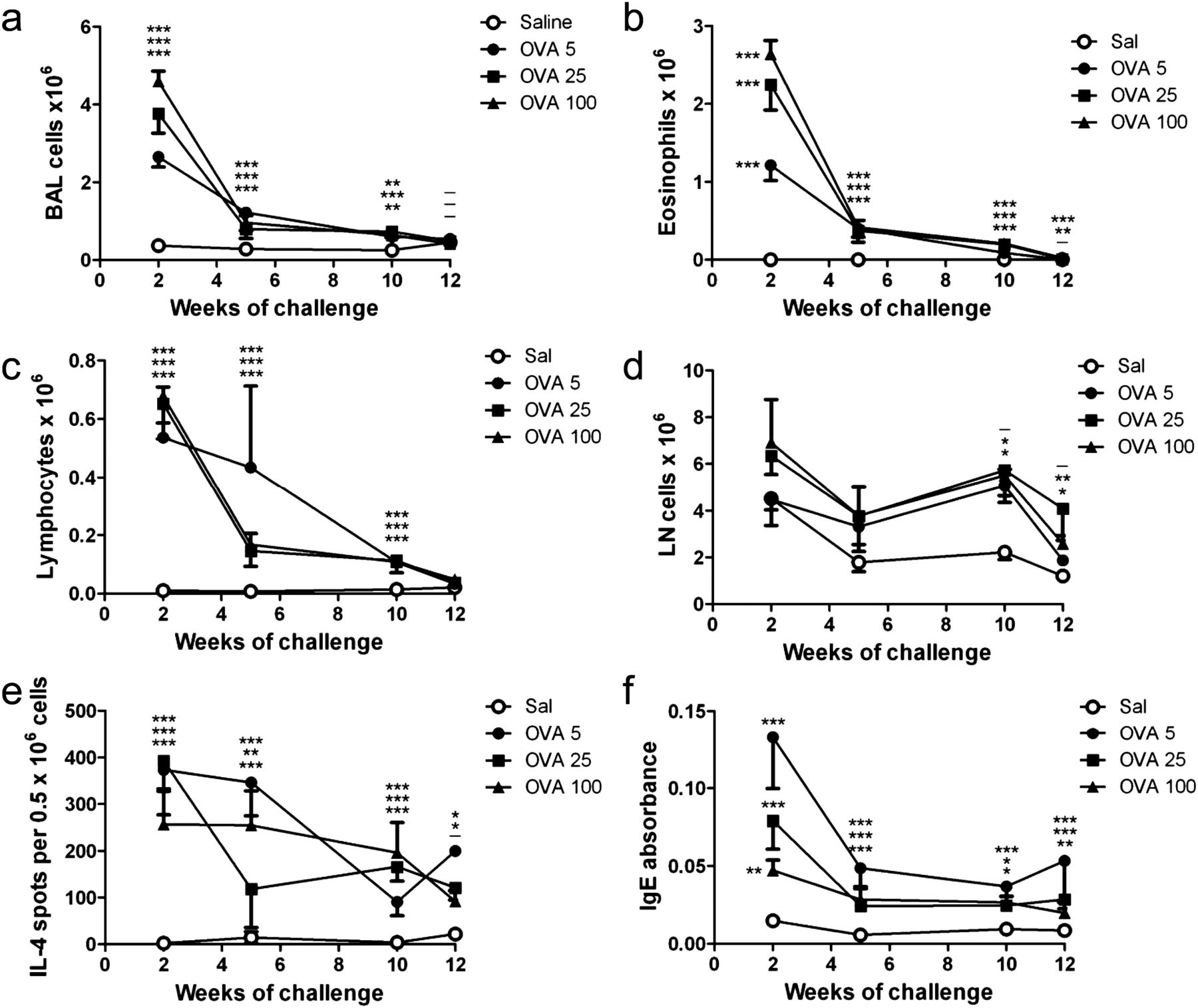
Chronic allergen challenge causes resolution of allergic inflammation independent of allergen challenge dose. Mice were sensitised and challenged with OVA as per online supplementary figure S1 A; controls received saline instead of OVA. (A–D) Counts of total bronchoalveolar lavage (BAL) cells, BAL eosinophils, BAL lymphocytes, and draining LN cells, and (E, F) frequency of interleukin (IL)-4-producing cells in the lung-draining LN, and levels of serum OVA-specific IgE were measured at the time points shown in (A). Mean ± SEM, n = 6–7 mice/group per time point. *p<0.05, **p<0.01, ***p<0.001; the order of symbols from top to bottom is OVA 5, 25 and 100 µg (relative to saline).
Allergen challenge increases BAL fluid levels of activin A, follistatin and TGF-β1
Next we determined the effect of chronic allergen challenge on concentrations of activin A, follistatin and TGF-β1 in the airways. Activin A and TGF-β1 concentrations were significantly elevated after 2 weeks of challenge, remaining high during allergen challenge, and dropping sharply once allergen challenge ceased (figure 2A,B). Follistatin concentrations showed a similar pattern, approaching control values by the 10-week time point (figure 2C). Thus, the elevated BAL fluid levels of activin A, TGF-β1 and follistatin seen during acute challenge are dependent on continued allergen challenge.

Activin A, TGF-β1 and follistatin levels are increased in bronchoalveolar lavage fluid during chronic allergen challenge. Mice were sensitised and challenged as described in online supplementary figure S1A, controls received saline instead of OVA. (A–C) Concentrations of activin A, TGF-β1 (ELISA) and follistatin (RIA) were measured at the time points shown in (A). Mean±SEM, n=6–7 mice/group per time point. *p<0.05, **p<0.01, ***p<0.001; the order of symbols from top to bottom is OVA 5, 25 and 100 µg (relative to saline).
Chronic allergen challenge decreases airway epithelial cell activin A and follistatin immunoreactivity, and induces airway remodelling
Airway epithelium in the saline group showed strong and uniform immunoreactivity for activin A and follistatin (figure 3A,C). In contrast, epithelial cell activin A and follistatin immunoreactivity were dramatically decreased during chronic allergen challenge, remaining low throughout the challenge period, and sharply increasing once allergen challenge ceased (figure 3B,D,E,F). After 5 weeks of challenge there was a significant increase in subepithelial collagen, consistent with other chronic challenge models,14–16 and this persisted at 10 weeks and after cessation of allergen challenge (figure 4A,B,E). The frequency of PAS-positive airway epithelial cells was significantly increased after 2 weeks of allergen challenge, remaining high throughout the challenge period, but declining sharply once allergen challenge stopped (figure 4C,D,F). Therefore, the increase in subepithelial collagen deposition persisted after cessation of allergen challenge, whereas mucus hypersecretion was dependent upon continued challenge. There was no consistent effect of allergen dose on the kinetics of these parameters.

Activin A and follistatin expression by airway epithelium in the normal lung is lost during chronic allergen challenge. Mice were sensitised and challenged as described in online supplementary figure S1A, controls received saline instead of OVA. Representative lung tissue sections stained immunohistochemically for activin A (A, B) and follistatin (C, D) from saline control (A, C) and OVA sensitised and challenged mice (B, D). (E, F) Frequency of activin A and follistatin immunoreactive cells per millimetre basement membrane (BM) as determined by quantitative image analysis. (A–D) Original magnification ×400. (E, F) Mean±SEM, n=6–7 mice/group per time point. *p<0.05, **p<0.01, ***p<0.001, respectively; order of symbols from top to bottom is OVA 5, 25 and 100 µg.

Chronic allergen challenge induces subepithelial collagen deposition and mucus hypersecretion. Mice were sensitised and challenged as described in online supplementary figure S1A, controls received saline instead of OVA. Representative lung tissue sections stained with Masson's Trichrome (A, B) or periodic acid Schiff (PAS) reagent (C, D) from saline control (A, C) and OVA sensitised and challenged mice (B, D). Collagen deposition in the subepithelial region (E) and frequency of PAS-positive airway epithelial cells per millimetre basement membrane (BM) (F) as determined by quantitative image analysis. (A–D) Original magnification ×400. (E, F) Mean±SEM, n=6–7 mice/group per time point. **p<0.01, ***p<0.001; order of symbols from top to bottom is OVA 5, 25 and 100 µg.
Follistatin administration during chronic allergen challenge attenuates Th2 cytokine production
To investigate the role of activin A in the remodelling process we instilled follistatin at the time of allergen challenge. We previously showed that 0.5–1 μg FS288 instilled prior to allergen challenge inhibits allergen-specific T helper 2 (Th2) cytokine and airway epithelial mucus production in an acute asthma model.6 Since activin A acts in a concentration-dependent manner19 (our own unpublished data), we tested the effect of 0.05, 0.5 and 5 μg follistatin instilled during chronic allergen challenge (see online supplementary figure S1B). Given that remodelling was independent of the OVA challenge dose (figure 4), we used 25 μg OVA per challenge for the remainder of our studies. Compared with mice challenged with OVA alone, low-dose follistatin significantly increased the number of BAL eosinophils (figure 5A) and total BAL cells (not shown). Surprisingly, however, higher follistatin doses had no effect on BAL eosinophils or total BAL cell counts. Follistatin dose-dependently increased lung-draining LN cell counts (figure 5B), but decreased the frequency of IL-4, IL-5 and IL-13 producing cells in the lung-draining LN (figure 5C–E). Thus, overall there was a marginal decrease in numbers of Th2 cytokine-producing cells (see online supplementary figure S2). In contrast, BAL fluid IL-13 concentrations were only significantly decreased in mice treated with low-dose follistatin (see online supplementary figure S3). We questioned whether the decreased frequency of Th2 cytokine-producing cells was mediated by regulatory T cells (Treg) or induction of a Th1-biased response. Lung CD4+CD25+Foxp3+ Treg numbers and proportions were increased by chronic allergen challenge, as expected,20 but were not further increased by follistatin treatment (see online supplementary figure S4). BAL fluid interferon γ levels were below the limit of detection in all treatment groups (data not shown). Follistatin instillation also dose-dependently decreased serum OVA-specific IgE levels (figure 5F). Thus follistatin exerts distinct dose-dependent effects in different pulmonary compartments, with a trend towards decreased frequency of Th2 cytokine-producing cells and serum OVA-specific IgE at the highest dose.

High-dose follistatin instillation during chronic allergen challenge inhibits allergen-specific T helper 2 cytokine production in the lung draining LN and serum allergen-specific IgE. Mice were sensitised as described in online supplementary figure S1B, and challenged with OVA, or OVA and follistatin; controls received saline instead of OVA or follistatin. Number of bronchoalveolar lavage eosinophils (A) and lung-draining LN cells (B). (C–E) Frequency of OVA-specific interleukin (IL)-4, IL-5 and IL-13 producing cells in the lung-draining LN. (F) Levels of serum OVA-specific IgE. Mean±SEM, n=6–8 mice/group. *p<0.05, ***p<0.001. FS, follistatin; Sal, saline.
Follistatin during chronic allergen challenge inhibits BAL fluid activin A and TGF-β1 concentrations, and airway remodelling
Follistatin instillation during chronic allergen challenge dose-dependently decreased BAL fluid activin A concentrations, with the 5 μg follistatin group decreased to approximately saline levels (figure 6A). Similarly, high-dose follistatin significantly decreased BAL fluid TGF-β1 concentrations (figure 6B). Notably, quantitative image analysis demonstrated that follistatin at all three doses significantly decreased collagen deposition in the subepithelial region (figure 6C). Furthermore, 5 μg follistatin significantly decreased the frequency of airway epithelial mucus-producing cells (figure 6D). Thus, follistatin inhibited airway activin A and TGF-β1 levels, and mucus hypersecretion and subepithelial collagen deposition.

Follistatin instillation during chronic allergen challenge inhibits bronchoalveolar lavage (BAL) fluid levels of activin A and TGF-β1, subepithelial collagen deposition and mucus hypersecretion. Mice were sensitised as described in online supplementary figure S1B, and challenged with OVA, or OVA and follistatin; controls received saline instead of OVA or follistatin. BAL fluid concentrations of activin A (A) and TGF- β1 (B). (C) Collagen deposition in the subepithelial region (D) and frequency of periodic acid Schiff (PAS)-positive airway epithelial cells per millimetre basement membrane as determined by quantitative image analysis. Mean±SEM, n=6–8 mice/group. *p<0.05, **p<0.01, ***p<0.001. FS, follistatin; Sal, saline.
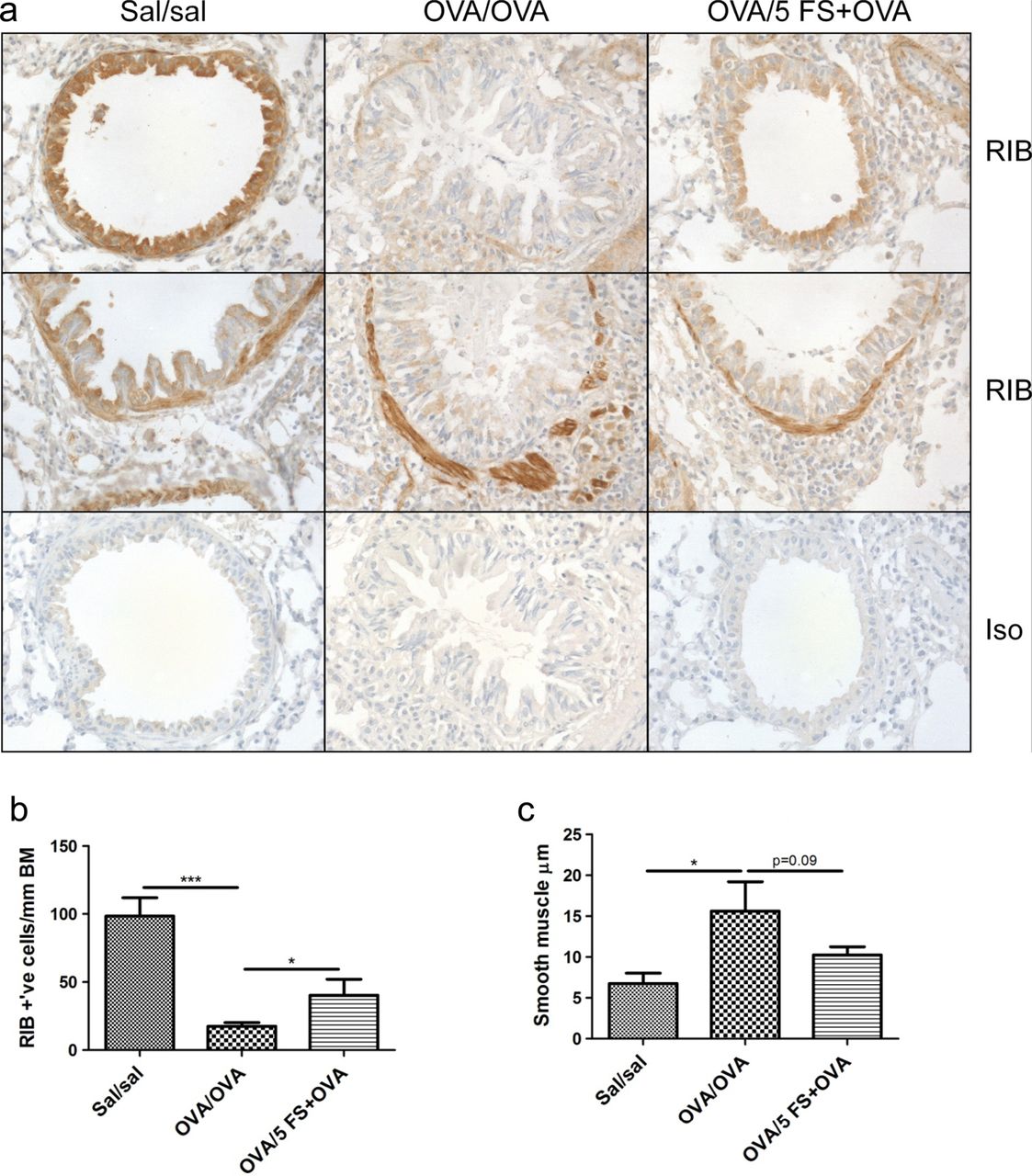
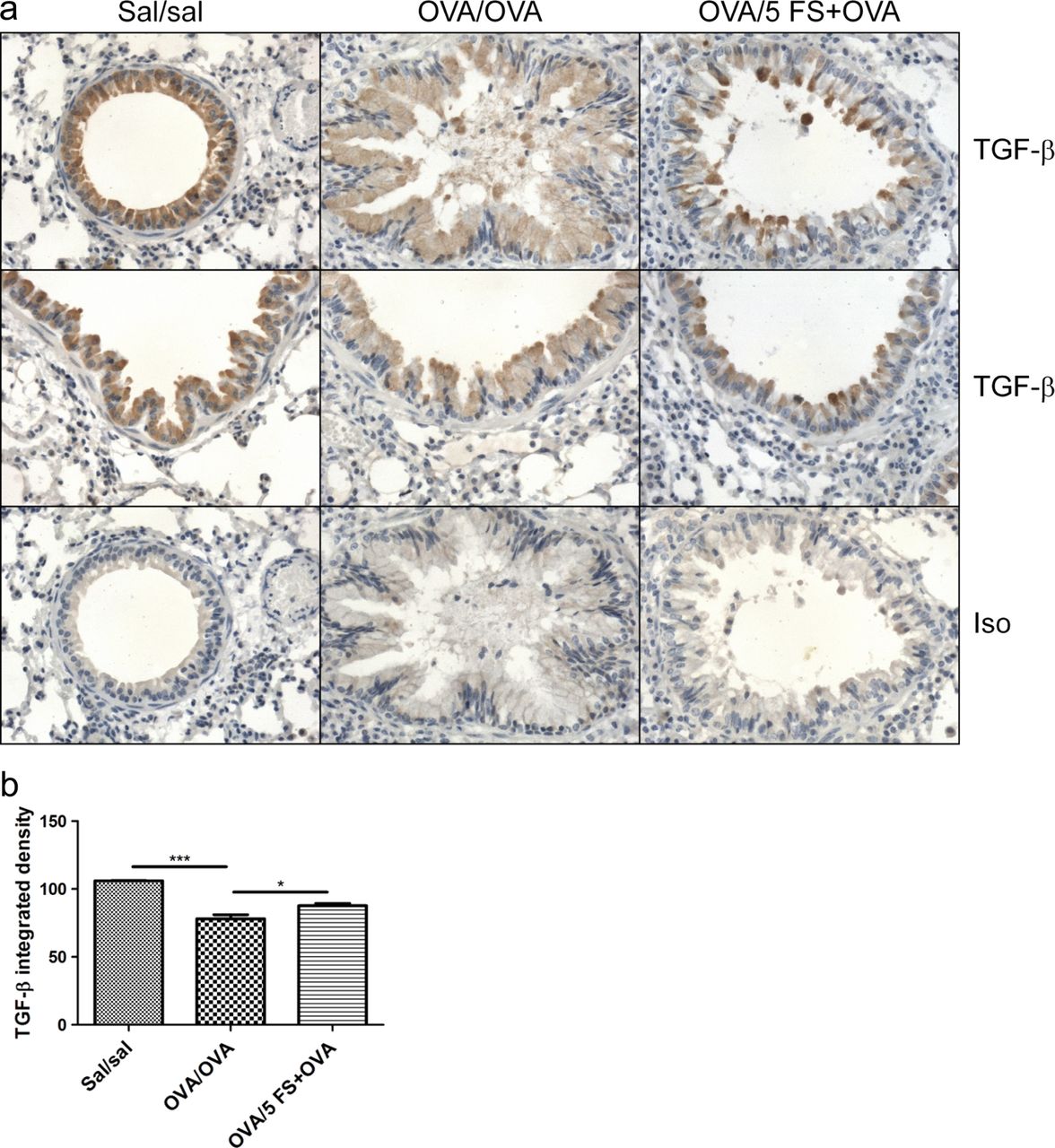
Follistatin inhibits the loss of airway epithelial TGF-β and ActRIB immunostaining during chronic allergen challenge
Immunohistochemistry showed strong TGF-β and ActRIB/ALK4 staining in normal (sal/sal) airway epithelium, which was significantly decreased in chronically challenged mice, and this decrease was significantly inhibited by follistatin (figures 7 and 8). There was strong immunostaining for ActRIB in the subepithelial smooth muscle of large airways in all groups. Subepithelial smooth muscle thickness was significantly increased more than twofold in OVA/OVA mice, and there was a strong trend for inhibition of this increase by 5 μg follistatin (p=0.09) (figure 8A,C). The strong immunoreactivity for activin A, ActRIIA and ActRIIB in normal airway epithelium mice was significantly decreased in chronically challenged mice, but this was not affected by follistatin treatment (see online supplementary figures S5 and S6). Apart from occasional activin A-positive macrophages in the lamina propria, there was weak and/or diffuse activin A, TGF-β and activin receptor staining of inflammatory cells in the OVA/OVA and OVA/5 FS+OVA groups.

Follistatin treatment during chronic allergen challenge diminishes the loss of TGF-β immunoreactivity in airway epithelium. Mice were sensitised as described in online supplementary figure S1B, and challenged with OVA, or OVA and follistatin; controls received saline instead of OVA or follistatin. (A) Formalin-fixed lung sections were stained immunohistochemically with antibody to TGF-β1. Representative micrographs, original magnification ×400. (B) Airway epithelial TGF-β staining intensity in small–medium airways. Mean±SEM, n=3 mice/group. *p<0.05, ***p<0.001. FS, follistatin; Sal, saline.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Follistatin treatment during chronic allergen challenge diminishes the loss of ActRIB/ALK4 immunoreactivity in airway epithelium. Mice were sensitised as described in online supplementary figure S1B, and challenged with OVA, or OVA and follistatin; controls received saline instead of OVA or follistatin. (A) Formalin-fixed lung sections were stained immunohistochemically with antibody to ActRIB. Representative micrographs, original magnification ×400. Frequency of ActRIB-positive airway epithelial cells in small–medium airways (B) and subepithelial muscle thickness in large airways (C). Mean±SEM, n=3 mice/group. *p<0.05, ***p<0.001. FS, follistatin; Sal, saline.
Discussion
In most patients stable asthma is well controlled on current medication but management of severe asthma remains inadequate. Despite the importance of inflammation in asthma pathogenesis, drugs targeting key inflammatory cells and inflammatory mediators show limited clinical benefit.1 These disappointing results suggested a reappraisal of asthma pathogenesis.1 Accumulating evidence indicates that activin A drives tissue fibrosis in a variety of organs,21–24 and is implicated in airway remodelling.12 ,13 In the current study we established a murine model of chronic allergic asthma to investigate the ability of follistatin to inhibit development of airway remodelling. Our results show that high-dose follistatin decreased airway activin A and TGF-β1 concentrations, and inhibited the decrease in TGF-β and ActRIB/ALK4 immunostaining observed in the airway epithelium of chronically challenged mice. Follistatin further inhibited mucus hypersecretion, subepithelial collagen deposition and thickening of the subepithelial smooth muscle, indicating a potential therapeutic role for follistatin in the prevention of airway remodelling.
Our data show that BAL fluid concentrations of activin A are elevated after 2 weeks of challenge, the earliest time point examined. Our previous studies show that increases in BAL fluid activin A concentrations occur within 24 h after allergen challenge,6 consistent with reports of increased activin A after one to three allergen challenges.10 ,13 The increased BAL fluid activin A and loss of activin A immunolocalisation in airway epithelium were rapidly reversed upon cessation of allergen challenge. These changes in airway and epithelial activin A expression mirrored the pattern seen for TGF-β1, consistent with other reports,14 ,25 suggesting release of activin A from epithelium into the airspaces. Similar changes were observed for follistatin, supporting the idea that follistatin is rapidly released as an endogenous antagonist of activin A bioactivity.6 ,26 One factor driving altered activin A compartmentalisation is IL-13,9 a key instigator of airway remodelling.15 ,27 ,28 Our observation that BAL fluid concentrations of TGF-β1 showed almost identical kinetics to activin A is not surprising since activin A and TGF-β1 stimulate secretion of one another,5 ,29–31 and IL-13 stimulates production of activin A and TGF-β1.9 ,27
Follistatin instillation during chronic allergen challenge decreased BAL fluid activin A and TGF-β1 concentrations. Since follistatin inhibited the loss of airway epithelial TGF-β during chronic challenge, the decreased BAL fluid TGF-β could simply reflect its decreased liberation from airway epithelium. The decreased BAL fluid activin A levels are likely due to formation of follistatin–activin A complexes. Subsequently, the decrease in available activin A would lead to decreased BAL fluid TGF-β1.5 ,29–31 Given the key role for TGF-β1 in airway remodelling,27 a simplistic interpretation would be that follistatin attenuated airway remodelling via inhibition of TGF-β1. However, follistatin binds to activin A with high affinity, but does not bind TGF-β1 or TGF-β2.32 ,33 Furthermore, activin A has been implicated in lung fibrosis via promotion of collagen and α-smooth muscle actin synthesis, and proliferation of human airway fibrobasts and smooth muscle,5 ,10 ,11 and follistatin decreases collagen secretion by pancreatic and hepatic stellate cells, and renal fibroblasts.29–31 Receptor-blocking experiments using dominant negative activin A and TGF-β type II receptors suggested that TGF-β action is partly mediated by secreted activin A.30 Together, these studies suggest that follistatin inhibits airway remodelling by blocking activin A, thereby inhibiting TGF-β1 production, positioning activin A as a central regulator of fibrosis.
We observed a general loss of activin receptor staining in airway epithelium following chronic allergen challenge. The thickened subepithelial smooth muscle showed strong ActRIB/ALK4 immunostaining but weak immunostaining for ActRIIA and ActRIIB. Subepithelial cells in the same location and with similar morphology stain with α-smooth muscle actin,16 emphasising that the ActRIB/ALK4-stained cells are smooth muscle or myofibroblasts. Similarly, decreased activin type I and II receptor expression was observed in airway epithelium and/or subepithelial cells in patients with asthma.7 In contrast, increased ALK4 and ActRIIA expression was found in airway epithelium of OVA-sensitised mice following acute allergen challenge,34 and increased ALK4 and ActRIIA expression was found in asthmatic airway epithelium 24 h after allergen challenge.8 Conceivably, differences in activin receptor expression between studies are due to different kinetics, with increased activin receptor expression occurring early after allergen challenge, decreasing at later times (ie, 5 weeks).
Chronic inflammation may be a driver of airway remodelling.1 However, several studies report dissociation between inflammation and airway remodelling. Neutralisation of TGF-β1 specifically, or all TGF-β isoforms, in OVA chronic allergen challenge models suppressed pulmonary fibrosis, without affecting airway inflammation, eosinophilia, or IL-5 or IL-13 production.25 ,35 Furthermore, overexpression of the activin/TGF-β1 signalling intermediate Smad2 in airway epithelium exacerbated airway remodelling in a house dust mite allergen challenge model.13 Intraperitoneal injection of activin A neutralising antibody markedly inhibited airway remodelling, but had no effect on lung inflammation, airway eosinophilia or Th2 cytokine production. Our data demonstrate that while follistatin dose-dependently modulated inflammation, the inhibition of subepithelial fibrosis was dose independent. However, BAL fluid activin A and TGF-β1 levels were only inhibited by high-dose follistatin, suggesting that even subtle decreases in local tissue concentrations of activin A and TGF-β1 can decrease fibrosis. Thus, while ELISA for activin A and TGF-β1 in the BAL fluid provides insight into lung levels of these mediators, it does not exactly mirror changes in mediator concentrations in the local tissue microenvironment.
Recently, antibody-mediated neutralisation of activin A during allergen challenge was demonstrated to exacerbate acute allergic airway inflammation.7 However, in our model of chronic allergic inflammation, inhibiting activin A with intermediate or high-dose intranasal follistatin suppressed OVA-specific IgE and allergen-specific Th2 cytokine production, and had no effect on airway eosinophilia. The attenuated Th2 cytokine production in the draining LN was not due to follistatin-induced expansion of CD4+CD25+Foxp3+ Treg in the lung, nor was it due to a switch to a Th1-type immune response. Thus, follistatin must exert this dampening effect via the induction of an alternative suppressive mechanism, such as the selective deletion/apoptosis of Th2 cells. These data suggest that the outcome of blocking activin A depends on the method (antibody vs follistatin) and/or the site (systemic vs local) of inhibition. Our finding that the lowest follistatin dose (0.05 μg) enhanced airway eosinophilia points to concentration-dependent effects of activin A on immune function. Analogous concentration-dependent morphogen effects of activin A occur during development.19
In summary, our study shows that follistatin inhibited activin A and TGF-β1 secretion into the airway lumen during chronic allergen challenge, and significantly decreased subepithelial fibrosis and airway epithelial mucus production. Follistatin treatment also caused a dose-dependent modulation of allergic airway inflammation, suggesting ‘morphogen-like’ effects of activin A on immune function. This is consistent with apparently contradictory observations of pro-inflammatory versus anti-inflammatory effects of activin A in other systems.2 Our study reinforces the idea that activin A is a key driver of inflammation and fibrosis, and indicates that follistatin represents an attractive potential therapeutic for the prevention of fibrosis in asthma.
Acknowledgments
The authors thank Professor Gary Anderson (University of Melbourne) for advice on establishing the mouse chronic asthma model and Ms Genevieve Tan (Department of Immunology, Monash University) for assistance with image analysis. We are grateful to Professor Jennifer Wilkinson-Berka and Pam Tong Zhu (Department of Immunology, Monash University) for advice with the TGF-β immunohistochemistry, and Elizabeth Richards (Department of Biochemistry and Molecular Biology, Monash University) for assistance with the activin A and activin receptor immunohistochemistry.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CLH wrote the paper, designed, performed and analysed experiments. HN designed, performed and analysed experiments, and produced rhFS288. RM performed and analysed experiments. JY performed experiments. DO analysed experiments. MP provided intellectual guidance. KL provided advice and helped with techniques. CH helped produce rhFS288. JM wrote the paper and provided intellectual guidance. ROH wrote the paper and provided intellectual guidance.
-
Funding Supported by grants from the National Health and Medical Research Council and the Co-operative Research Centre for Asthma and Airways, Australia.
-
Competing interests None.
-
Ethics approval This study was conducted with the approval of the Alfred Medical Research and Education Precinct Animal Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.