Article Text

Statistics from Altmetric.com
- Bacterial infection
- cystic fibrosis
- innate immunity
- neutrophil biology
- opportunist lung infections
- oxidative stress
- respiratory infection
- tuberculosis airway epithelium
- bronchoscopy
- paediatric lung disease
- paediatric physician
Pseudomonas aeruginosa is the most frequently encountered lung pathogen in patients with cystic fibrosis (CF). Following initial, often intermittent, episodes of infection, it becomes a permanently established component of the chronically infected lung in more than 80% of patients and confers an adverse prognosis. The predisposition of the CF airway to P aeruginosa is incompletely understood but our current concept of the sequence of events leading from initial acquisition of infection through to the chronic state with severe but often ineffective inflammation is becoming clearer.
Initial infection
The defect in ion transport resulting from abnormal CF transmembrane conductance regulator protein leads to a dehydrated airway surface. Mucociliary clearance suffers and either shed bacterial components, such as flagella, or, more controversially, the bacteria themselves, trigger inflammatory responses via direct contact with cell surface glycoproteins. Increased expression of pro-inflammatory cytokines leads to neutrophil influx from the systemic circulation. Although at early stages of infection P aeruginosa can be successfully eradicated from the airway, the CF inflammatory response has been shown to be both exaggerated (out of proportion with the bacterial load) and prolonged (failing to be successfully regulated by the usual ‘switch-off’ signals such as lipoxins); both likely contribute to the destructive changes in the airway wall characteristic of the disease. If the initial infecting bacteria survive, they lose their flagella, becoming thereby less detectable to the host immune system. They enter a sessile state and reproduce making use of the high concentrations of amino acids in the sputum to extremely high bacterial densities (often ≥109 colony-forming units ml−1). They appear to be attracted to the hypoxic environment in CF mucus plugs and this hypoxia, and other environmental conditions, may encourage a number of genetic and phenotypic switches as the bacteria adapt to the chronically infecting state.
Host defence and the evasion tactics used by P aeruginosa
Phagocytosis, long considered the major neutrophil defence mechanism, is impaired in CF related to: a) the primary CF transmembrane conductance regulator deficiency leading to abnormal intracellular HOCl concentrations and b) high airway luminal levels of proteases, which cleave the essential recognition molecules from the neutrophil surface.1 More recently, a method of extracellular killing has been recognised whereby both nuclear and mitochondrial DNA is extruded from the neutrophil and forms a ‘trap’ for bacteria. Bacterial killing results from the close juxtaposition on these chromatin strands of the bacteria and neutrophil granules containing elastase, cathepsins, defensins and other toxic enzymes. Abundant neutrophil extracellular traps (NETs) have been demonstrated in CF sputum; these contribute to sputum viscosity and can be degraded by rhDNase. NETosis was in fact the main mechanism of bacterial killing in the report of a recent, novel experiment designed to mimic the airway, with constant motion and mild sheer forces.2 This was in contrast to the static cultures almost universally employed for this type of investigation, in which phagocytosis predominates. NET-mediated killing was present even at low bacterial concentrations and with no prior requirement for opsonisation. Importantly, this mechanism has been shown to be intact in CF neutrophils, which are able to trap and kill P aeruginosa organisms found at the early stages of infection. Interestingly, isolates from the same patients at later stages of chronic infection were resistant to this form of killing; the reasons for this remain incompletely understood, although mucoidy alone was found not to be the explanation. This might suggest that NET formation is important in the early stages of infection, but that at later stages, the bacteria adapts to resist this line of attack. Whether the mechanism(s) through which it is achieving this relates to any of the known immunoevasive mechanisms discussed below, or to new pathways, remains to be investigated.
P aeruginosa uses a number of strategies for immune evasion with virulence factors such as exotoxin A, haemolysin, phospholipase C, pyocyanin, hydrogen cyanide and alginate.3 Complement evasion is important during the initial stage of infection; alkaline protease and elastase degrade the complement activation product, C3b, deposited on the bacterial surface and lipopolysacharide variants interfere with C3b surface deposition. Alginate production following the mucoid switch will limit access of host factors such as complement as well as phagocytosis and access of antibodies to the bacterial cell: a factor that will increase in importance during chronic infection. Alginate increases the respiratory burst in neutrophils and may stimulate monocytes to produce pro-inflammatory cytokines.4
P aeruginosa engages in redox warfare with the innate immune system. The host attack on P aeruginosa has a strong redox component as neutrophils produce reactive oxygen species from their respiratory burst and epithelial cells produce the oxidant, hypothiocyanite (OSCN−), via the DUOX/LPO/SCN− system. P aeruginosa counterattacks with the virulence factor, pyocyanin (responsible for the green colour of P aeruginosa infected sputum), which competitively inhibits DUOX activity and generates superoxide.5 Along with the rhamnolipid detergents it induces necrosis and hence removal of neutrophils. This results in the release of tissue-damaging neutrophil products including myeloperoxidase, elastase, matrix metalloproteinase-9 and DNA; clearance of infection is unsuccessful while significant tissue damage ensues.
Genetic adaption: chronic infection, quorum sensing and biofilm formation
An increasing body of evidence supports the concept that the CF airway is a spatially and temporally heterogeneous environment that provides selective pressures for adaptive radiation and niche specialisation of infecting organisms. The appearance of colony morphology variants in the sputum is evidence of this. The most commonly recognised adaptation is the switch to a mucoid phenotype mentioned above. This is due to mutation of the antisigma factor encoding mucA gene that results in overproduction of the exopolysacharide alginate. Also frequently encountered are mutations in the lasR gene that encodes the regulator of N-(3-oxododecanoyl) homoserine-L-lactone (3-oxo-C12-HSL) quorum sensing system that sits atop of the complex cell–cell communication hierarchy in P aeruginosa. This leads to downregulation of virulence factor production (elastase, alkaline protease, rhamnolipids, pyocyanin and hydrogen cyanide, etc) during chronic infection. Mutations leading to antibiotic resistance start to appear (eg, mexZ mutations in the regulator of the MexXY-OprM multidrug-efflux pump) as do alterations in lipopolysacharide, loss of type 3 secretion systems and hypermutatbility. These adaptations generate a genetically and phenotypically heterogeneous population of P aeruginosa.3 Sublethal concentrations of antibiotics have been shown to increase the mutation frequency in bacteria and may be a currently unrecognised influence on the rate of adaptive radiation of P aeruginosa in the lung. Furthermore, the innate heterogeneity of the CF lung environment may encourage inducible resistance to antibiotics as well as antibiotic persister cells in the population. Either non-mucoid or mucoid organisms may develop into biofilm-like colonial aggregates (figure 1) via the process of quorum sensing. The production of highly membrane permeable molecules such as N-acylhomoserine lactones allow organisms to ‘sense’ neighbouring counterparts; once the concentration of these indicates that a quorum has been reached, switches occur in bacterial gene expression which lead to production of the biofilm matrix.3 Evidence that P aeruginosa forms biofilms in the CF airway comes from the presence of quorum sensing molecules in sputum. However, most knowledge of biofilm physiology comes from studying abiotic surfaces in vitro and care has to be taken extrapolating such data to the CF lung environment where bacterial populations appear to exist in sputum surrounded by neutrophils rather than adhering to epithelial surfaces. The macrolide antibiotic, azithromycin, has significant clinical benefit in patients with CF despite not possessing antipseudomonal properties; evidence suggests that it may however be able to kill bacteria living in biofilms. Other groups are exploring the therapeutic potential of agents which directly break down biofilm matrix allowing access to the bacteria by conventional antimicrobial agents, an aim supported by the fact that plant species naturally producing acylhomoserine lactone-degrading enzymes are inherently resistant to chronic infection with this pathogen.
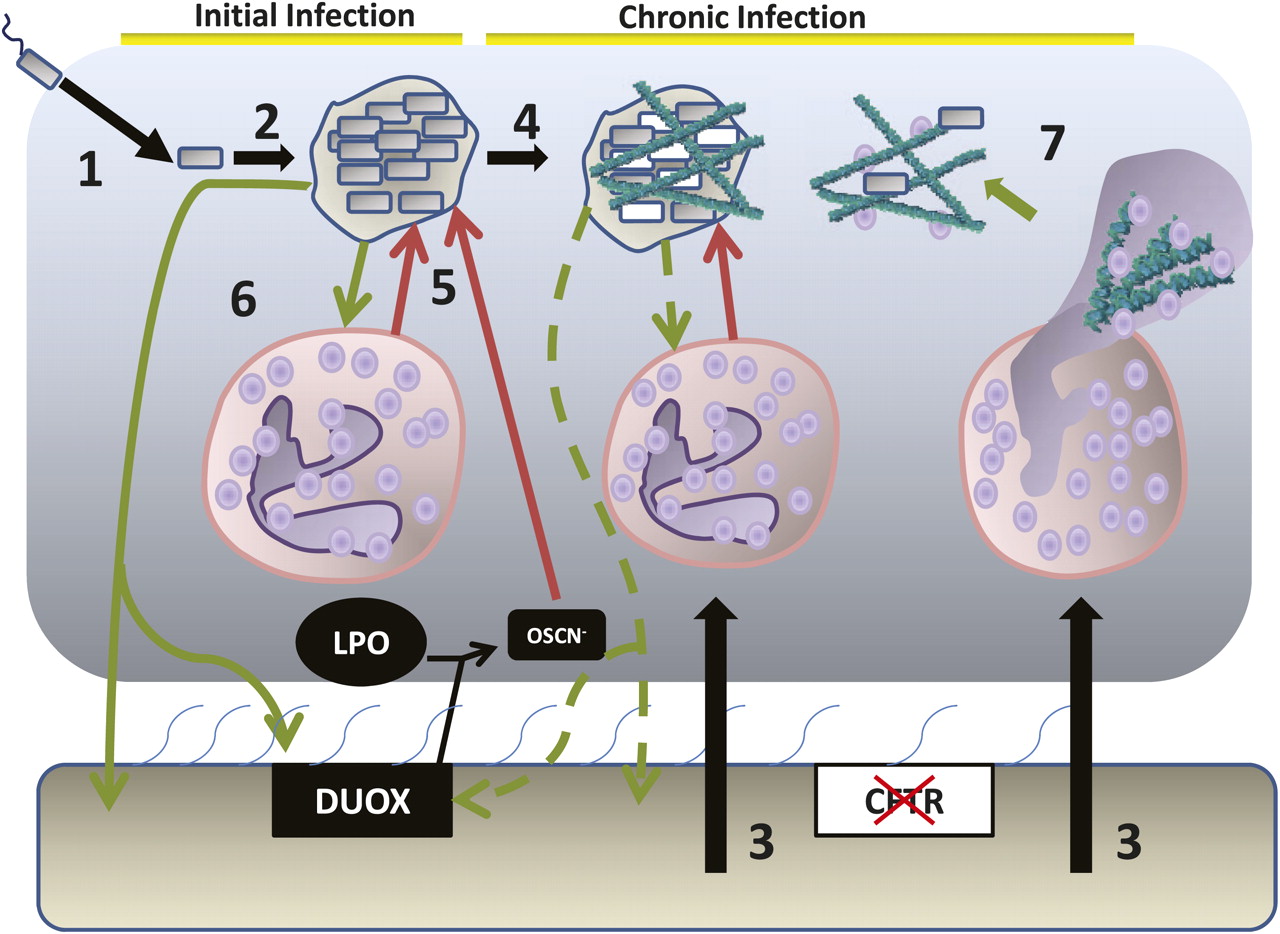
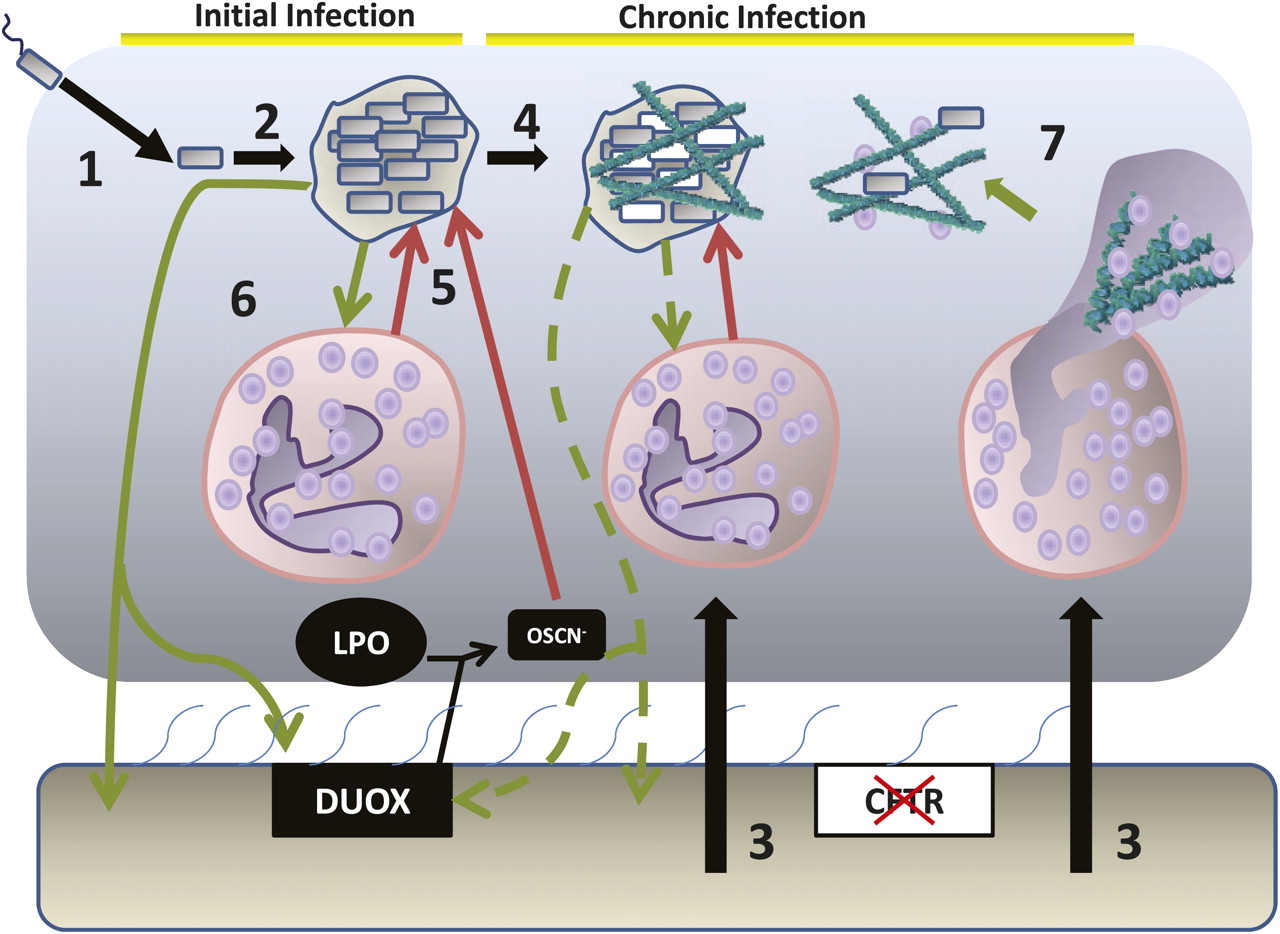{kind=link}
The epithelial environment of the cystic fibrosis lung and the stages of infection with Pseudomonas aeruginosa: 1. Initial infection with P aeruginosa 2. Growth and formation of dense biofilm-like colonial aggregates of bacteria. 3. Massive neutrophil influx into the airway surface environment. 4. Bacterial adaptation and evolution to the chronic infection state, including the switch to mucoid P aeruginosa (white cells) and downregulation of virulence factor production. 5. The host attacks but fails to resolve the infection leading to collateral damage to surrounding tissues as the activated neutrophils release elastase, collagenase and the reactive oxygen species that result from the neutrophil respiratory burst. 6. Bacterial counterattack through virulence factor production, including compounds such as pyocyanin, which attack the LPO/DUOX and with rhamnolipids lead to neutrophil necrosis. Alginate formation supports the biofilm matrix. 7. Neutrophil NETosis leads to production of neutrophil extracellular traps that can kill bacteria but also release DNA that can provide structural support for the P aeruginosa biofilm matrix and increase the viscosity of the mucus, further reducing mucociliatory clearance.
In summary, the airway becomes infected with P aeruginosa early in the life of most CF patients. Despite the success of early eradication attempts, infection will become chronic in the majority of these, aided by the organisms' ability to evade and disarm the host immune system. Our increasing understanding of the mechanisms involved, both bacterial and host, could, in future, lead to the identification of new drug targets to prevent the inevitable and irreversible airway damage that results.
Footnotes
Competing interests None.
Provenance and peer review Commissioned; internally peer reviewed.