Article Text

Abstract
The composition of the lung microbiome contributes to both health and disease, including obstructive lung disease. Because it has been estimated that over 70% of the bacterial species on body surfaces cannot be cultured by currently available techniques, traditional culture techniques are no longer the gold standard for microbial investigation. Advanced techniques that identify bacterial sequences, including the 16S ribosomal RNA gene, have provided new insights into the depth and breadth of microbiota present both in the diseased and normal lung. In asthma, the composition of the microbiome of the lung and gut during early childhood development may play a key role in the development of asthma, while specific airway microbiota are associated with chronic asthma in adults. Early bacterial stimulation appears to reduce asthma susceptibility by helping the immune system develop lifelong tolerance to innocuous antigens. By contrast, perturbations in the microbiome from antibiotic use may increase the risk for asthma development. In chronic obstructive pulmonary disease, bacterial colonisation has been associated with a chronic bronchitic phenotype, increased risk of exacerbations, and accelerated loss of lung function. In cystic fibrosis, studies utilising culture-independent methods have identified associations between decreased bacterial community diversity and reduced lung function; colonisation with Pseudomonas aeruginosa has been associated with the presence of certain CFTR mutations. Genomic analysis of the lung microbiome is a young field, but has the potential to define the relationship between lung microbiome composition and disease course. Whether we can manipulate bacterial communities to improve clinical outcomes remains to be seen.
- COPD mechanisms
- cystic fibrosis
- asthma
- bacterial infection
- COPD epidemiology
- COPD exacerbations
- emphysema
- interstitial fibrosis
- long term oxygen therapy (LTOT)
- lung transplantation
- lung volume reduction surgery
- pulmonary rehabilitation
- asthma guidelines
- asthma mechanisms
- perception of asthma/breathlessness
- respiratory infection
- viral infection
- allergic lung disease
- COPD pathology
- cytokine biology
- innate immunity
- lymphocyte biology
- macrophage biology
- tobacco and the lung
- viral infection
- COPD pharmacology
- pneumonia
- pneumonia
- asthma epidemiology
- asthma genetics
- lung physiology
Statistics from Altmetric.com
- COPD mechanisms
- cystic fibrosis
- asthma
- bacterial infection
- COPD epidemiology
- COPD exacerbations
- emphysema
- interstitial fibrosis
- long term oxygen therapy (LTOT)
- lung transplantation
- lung volume reduction surgery
- pulmonary rehabilitation
- asthma guidelines
- asthma mechanisms
- perception of asthma/breathlessness
- respiratory infection
- viral infection
- allergic lung disease
- COPD pathology
- cytokine biology
- innate immunity
- lymphocyte biology
- macrophage biology
- tobacco and the lung
- viral infection
- COPD pharmacology
- pneumonia
- pneumonia
- asthma epidemiology
- asthma genetics
- lung physiology
Introduction
Although it is a long-held belief that the normal human lung is sterile, increasing evidence suggests that it is not, and that the composition of the lung microbiome (the entire community of microorganisms that inhabit the lower respiratory tract) contributes centrally to both health and disease, particularly obstructive lung disease.1 2 Despite the large number of bacterial species that have been grown from human mucosal sites, it is estimated that over 70% of the bacterial species inhabiting bodily surfaces cannot be cultured by currently available techniques3 4 and that many of the remaining 20–30% of species are very difficult to culture.5 Hence, traditional culture techniques can no longer be considered the gold standard methodology for microbial investigation of the lung. The ability to identify microorganisms in a complex community that are sampled directly from their environment and identified based on unique genetic signatures has revolutionised our understanding of the indigenous microbes that inhabit virtually every organ in the body in communication with the external environment.6 In particular, techniques that identify bacteria on the basis of ribosomal RNA (rRNA) sequences are providing new insights into the depth and breadth of microbiota present both in the normal and diseased lung. The goal of this review is to introduce pulmonary clinicians and researchers to this revolution.
Methodology overview
This approach to studying the unseen microbial world originated over 30 years ago with the pioneering work of Carl Woese, who used rRNA sequence comparisons to reconstruct the evolutionary history of microbes.7–9 Norman Pace and colleagues subsequently built molecular tools for assessing the ecology of microorganisms.10 11 Since these early initial studies, more than 100 novel bacterial phyla (see figure 1) have been identified, most of which include currently unculturable representatives.12 The majority of culture-independent techniques are based on the PCR amplification of the 16S rRNA gene, a highly conserved locus of the bacterial genome (see table 1 for Glossary of Terms). Despite being relatively small (approximately 1.5 Kbp), the 16S rRNA gene possesses nine hypervariable regions that can facilitate genus and species identification.13 Amplification of conserved sequence stretches within the 16S rRNA gene, flanking these hypervariable regions, allow for the design of broad bacterial kingdom-specific primers. With these primers, amplicons of the 16S rRNA genes in a bacterial population can then be created and the sequence differences used to identify specific bacteria. Current molecular techniques can create entire microbial community fingerprints, locate and quantify bacteria geographically within the mucosa, quantify and estimate relative bacterial abundance and identify the members of the community. It is also possible to consider the microbial population as a whole ‘metagenome’ by applying high-throughput shot-gun sequencing to the entire population, that is, metagenomics, to identify the community members present and their genetically encoded functional capacity.

Bacterial classification system using Escherichia coli as an example.
Glossary of terms
Commonly employed techniques
By far, the most commonly used technique to measure bacterial abundance is real-time quantitative PCR (qPCR), which through the use of target-specific primers, can provide specificity at the genus level (see figure 1). Data can be expressed (and normalised) as total bacterial gene copy number per unit sample or per single copy host gene, if biopsy or other tissue samples are analysed. Another technique, fluorescent in situ hybridisation (FISH) uses fluorescent oligonucleotide probes that bind to a specific target sequence, that is, 16S rRNA, within the bacterial cell; after hybridisation to the target gene, the spatial distribution and number of target organisms is determined using a fluorescent microscope. In this way, FISH can be used to visualise and quantify abundance of bacteria in situ in their particular environments within the mucosal respiratory tract. These techniques have also been adapted for flow cytometry, permitting identification of individual bacteria.14 Universal bacterial probes and those for specific genera are available14; in the gastrointestinal tract, it is estimated that around 90% of bacteria can be labelled by some types of FISH probes.15
Molecular fingerprinting methods, denaturing and temperature gradient gel electrophoresis and terminal restriction fragment length polymorphisms (T-RFLP), have historically represented the quickest and simplest techniques for determining shifts in community structure and composition.14 T-RFLP involves amplification of 16S rRNA using a fluorescently labelled primer, digestion of the amplicons by a restriction enzyme, and separation of the digested fragments on an automated DNA sequencer. Finally, electropherograms showing the relative abundance of the individual fragments (T-RFs) are created.16 17 Changes in the community can be detected by the loss or gain of T-RFs from the profile. Various multi-dimensional statistics can be applied to these datasets, but definitive taxonomic assignments of T-RFs are usually not possible when analysing complex, highly variable communities.
Constructing libraries of 16S rRNA gene amplicons from a mixed bacterial population has been a useful method to identify community membership. Amplicons are cloned into a bacterial expression vector, the clones (colonies) are picked and their inserts sequenced (typically by the Sanger technique) to create a quantitative measurement. Sequences of the clones can be compared using algorithms that are classifier based or operational taxonomic unit (OTU) based; in either case, the output of such algorithms is linked to existing taxonomic databases, for example, the Ribosomal Database Project (RDP, http://rdp.cme.msu.edu/). The construction and analysis of these ‘clone libraries’ to study a bacterial community sample is a robust technique but suffers from the limitations of cloning bias, cost and depth of sampling.
To circumvent these limitations of clone library analysis, high-throughput sequencing of 16S rRNA gene amplicons can be utilised. The commonly used 454-pyrosequencing platform offers advantages due to sequence read lengths of over 400 bp (sufficient to cover at least two adjacent 16S rRNA hypervariable regions) and output of more than 106 high-quality sequence reads,18–20 permitting a more robust sampling of microbial communities.2 21–25 This platform also provides the benefit of permitting simultaneous analyses of multiple samples, through the use of barcodes or ‘tags’ that are attached to 16S rRNA amplicon pools arising from different samples.20 However, as sequencing technology advances, next-generation platforms are being adopted for microbial ecology studies and promise to revolutionise the field. Examples include Illumina (Illumina Corporation, San Diego, California, USA), which can produce up to 6×109 reads per run, or the PacBio RS system (Pacific Bioscenses, Menlo Park, California, USA), which promises even greater read depth using technology that detects single molecule sequencing reactions in real time.
Another approach, particularly useful for analysis of comparative bacterial community composition, is the 16S rRNA PhyloChip (designed at Lawrence Berkeley National Laboratory, manufactured by Affymetrix Corporation, Santa Clara, California, USA).26 27 The current G3 generation of this high-density phylogenetic microarray contains approximately 1.2 million probes that can differentiate approximately 60 000 bacterial taxa (defined as groups of organisms sharing ≥99% 16S rRNA sequence homology) and demonstrates excellent resolution of complex bacterial communities. The PhyloChip is particularly useful for identification of less abundant bacterial community members and their association with disease. Collectively, these culture-independent methods have begun to highlight a microbial world not previously recognised in the lungs.
Advantages and limitations of specific molecular techniques
This panoply of techniques should be appreciated as rapidly evolving and complementary; none is ideal for all research questions. For example, those based on amplified 16S RNA will detect only bacteria, whereas bacteria, fungi and viruses can be detected using shotgun sequencing approaches of total nucleic acid (DNA or RNA) that provide extensive depth of community coverage via enormous numbers of sequence reads, such as the Illumina platform. Although all are quantifiable in principal, some (FISH on tissue sections and the three types of molecular fingerprinting) do not readily lend themselves to quantitative results. Moreover, because the majority of these methods rely on analyses of DNA, there is some concern that they could detect non-viable organisms, although that issue is clearly less relevant when analysing RNA by these approaches, which originates from viable organisms and is inherently quite labile.
Additionally, all of these molecular techniques currently require extensive computational analysis. The time required to perform these analyses can exceed the time required to produce the raw data by an order of magnitude. Duration of data analysis is least for hybridisation-based techniques such as the PhyloChip, or for those typically analysing a restricted spectrum of specific organisms, such as FISH. By contrast, the enormous mass of data produced by sequencing techniques (454-pyrosequencing, and to an even greater extent, the Illumina and PacBio systems) can require weeks of computationally intensive analysis that is currently only partially amenable to automation. Nevertheless, as bioinformatics procedures advance and the cost of sequencing falls, it is foreseeable that these approaches could be incorporated into routine clinical practice.
A significant concern when such highly sensitive techniques as these are used to characterise microbial communities is the possibility of contamination during sample acquisition.28 This concern is particularly relevant for the lungs, given the necessity of traversing the upper respiratory tract which is intensely colonised by microorganisms. Data being collected from ongoing studies including the National Heart, Lung and Blood Institute (NHLBI) Lung Human Microbiome Project will help address these and other technical considerations about pulmonary sampling techniques for the lung microbiome.
It is reasonable to ask, what do these novel methodologies add to conventional culture results? For the practice of pulmonary medicine in 2012, the answer would have to be nothing, yet. None of these techniques have reached the stage that they can be ordered from a clinical laboratory to guide care of an individual patient. The next section, however, describes how research in the gastrointestinal tract has already radically changed thinking on host–microbiome interactions. With ever expanding data on the airway microbiota, it is only a matter of time before the composition and dynamics of microbiota in this niche are afforded the same level of investigation, and relationships between airway microbial assemblages and pulmonary health are identified.
Interactions between the microbiome and mucosal immunity
The most thoroughly studied mucosal microbiome in humans and mice is the gut,29–31 which provides lessons for the much more recently identified lung microbiome. The composition of the gut microbiome is remarkably stable within individuals32 and largely shared between healthy individuals,33 and yet it is sensitive to diet and powerfully able to impact host metabolism.34 35 An example is the strong correlation between the composition of the gut microbiota and obesity,36 37 apparently at least in part due to increased efficiency of caloric delivery to the host.38 39
Two features of the host–gut microbiome interaction are key to understanding why recognition of the lung microbiome is such an important concept for obstructive lung diseases. First, studies performed in mice show that the gut microbiome is essential to shaping the host immune system. By controlling the activation state of gut dendritic cells, a single intestinal organism, segmented filamentous bacteria, regulates the systemic balance of T-helper type 17 cells and T-regulatory cells in mice, and thereby the outcome of subsequent pathogen infection.40 41 This is likely to be only one of many examples of immunoregulation by commensal bacteria.42–46
Second, gut microbiota appear to play an integral facilitating role in inflammatory bowel diseases. Although there have been suggestions that specific pathogens contribute to inflammatory bowel diseases, the more accepted current theory is that gut inflammation is driven by an abnormal host response to bacterial antigens to which the host is normally tolerant.47 Both Crohn's disease and ulcerative colitis patients show reductions in the numbers and diversity of members of the mucosa-associated phyla Firmicutes and Bacteroidetes.48 Analogous changes have recently been identified in asthma and chronic obstructive pulmonary disease (COPD).1 2 Of course, in all these diseases, separating cause and effect will be difficult, but it is intriguing that beneficial effects of chronic azithromycin therapy have been shown in asthma (reviewed in Martinez et al49), cystic fibrosis (CF)50 and COPD,51 although in CF, benefit is limited to those with Pseudomonas colonisation. Azithromycin combines antibacterial, anti-inflammatory and immunomodulatory effects that could plausibly alter the composition of bacterial communities in the lungs.
Obstructive disorders
Microbiome in asthma
The role of microbial colonisation or infection by specific organisms in the pathogenesis of asthma has been much less studied than its role in allergic sensitisation and challenge. An increasing body of evidence, however, suggests that the composition of the microbiome of the gut and/or airway may be important in determining the risk of asthma development and contribute to chronic stable asthma.
Bacterial exposures in early childhood are postulated to diminish the risk of subsequent asthma or allergy, known as the ‘hygiene hypothesis’.52 Since this concept was first proposed, research findings have broadened the scope of potential microbial exposures that must be considered. Rather than infection per se, exposure to and perhaps colonisation by specific microbiota during infancy are regarded as important in protection against asthma or allergy. Environmental exposures that appear to be protective against asthma or allergic sensitisation include childhood contact with pet dogs53 and exposure to farm animals.54 55 That microbiologic mechanisms are likely involved in these epidemiologic observations is indicated by the findings that higher endotoxin load in bedding and a greater diversity of environmental microorganism exposures during childhood are associated with lower rates of asthma or atopy.54 55 Moreover, Fujimura and colleagues recently demonstrated that homes with a pet that was permitted both indoors and outdoors possessed house dust with a significantly greater diversity of bacteria and fewer fungal types,56 fuelling speculation that differential infant gastrointestinal colonisation patterns associated with such exposures may provide protection against allergic disease development.
It is currently hypothesised that such environmental exposures shape the risks of developing asthma or atopy by altering the composition of the human gut and airway microbiota initially acquired during infancy. Commensal gut microbiota are arguably our most important source of microbial stimulation. They are key in shaping early immunologic development and, in turn, susceptibility to inflammatory and allergic diseases.57 Factors that can impact gut microbiota composition in early life and which have been associated with an increased risk of asthma or allergy include formula feeding, birth by caesarean section, and early antibiotic administration.58 59 The results of studies that have analysed stool microbiota indicate that these risk factors are associated with differences in gut microbiota composition. Infants born by caesarean section and those fed formula are more often colonised by Clostridium difficile, compared with infants born by vaginal delivery or those who are breastfed, respectively.58 Counts of Clostridia are higher and counts of Bifidobacteria are lower in stool samples from infants who subsequently developed atopic manifestations.60–62
Recent studies of the airway microbiome have demonstrated that multiple types of bacteria are linked with asthma development and chronic stable disease.1 63 64 In a prospective study of 321 Danish infants at risk for asthma, culture-based detection of Streptococcus pneumoniae, Moraxella catarrhalis, Haemophilus influenzae or a combination of these organisms in hypopharyngeal samples at 1 month of age was associated with an increased prevalence of asthma at age 5 years (33% colonised vs 10% non-colonised).63 Peripheral eosinophilia and elevated total IgE were also associated with these organisms, suggesting that the composition of the airway microbiota acquired in infancy may influence susceptibility to allergic risk, as observed with gut microbiota, although further research is needed. More recently, two culture-independent studies of the lower airway microbiota in chronic asthma using 16S rRNA gene sequence analysis1 64 demonstrated the presence of a richer microbial community in the lower airways than previously recognised. In a study of 24 adults and 20 children, pathogenic members of the Proteobacteria phylum, in particular Haemophilus spp., were more frequently identified in bronchial airway samples from adults with asthma or COPD and children with difficult asthma, compared with controls.1 Although all patients with asthma in this study had been prescribed corticosteroid therapies, confounding any determination of causation, the findings suggest that a specific composition of airway microbiota may characterise patients with chronic asthma. Similarly, differences in the airway microbiota between healthy controls and patients with asthma on inhaled corticosteroid therapy were observed in a larger study of 75 adults, in whom 16S rRNA-based microarray analysis of protected bronchial brushings was performed.64 Patients with asthma demonstrated significantly higher bacterial diversity and greater bacterial burden compared with healthy controls. Moreover, airway microbiota composition and greater bacterial diversity were significantly correlated with bronchial hyperresponsiveness, including the relative abundance of specific microbiota belonging to bacterial families within the Proteobacteria. Further research is needed to understand functional aspects of the airway microbiome and how they may contribute to pathophysiologic processes in asthma. Collectively, the results from these studies invite greater consideration for the role of a polymicrobial airway community in the pathogenesis of asthma.
Microbiome in the smoking lung and COPD
COPD is a highly prevalent disorder characterised by airflow obstruction that is not completely reversible65 and which exhibits increasing mortality and morbidity.66 67 COPD is also known to be a highly heterogeneous disease, with widely varying clinical symptoms, health status and disease progression.68 COPD is clearly an inflammatory disorder; the inflammatory/immune response persists despite smoking cessation, varies by the patient population, method of assessment, and timing of measurement.69 In addition, during episodes of increased symptoms, termed acute exacerbations of COPD (AECOPD), inflammation increases.69 AECOPD have been associated with impaired health status, physiological decline and increased mortality. The genesis for the persistent inflammatory/immune response in COPD remains controversial; one theory includes perpetuation of an immune response to auto-antigens initially resulting from inflammatory and oxidative lung injury.70 Persistent or recurrent infection may play a particularly crucial role, as infection may serve as a stimulator of specific immune responses or as a polyclonal activator. Bacterial colonisation (determined by traditional culture-based methodologies) has been associated with a chronic bronchitic phenotype, with increased risk of exacerbations, and with accelerated loss of lung function.69 Recent data support a similar process with the genesis of emphysema and airway structural abnormality.69 71
Chronic bacterial colonisation in patients with COPD has been traditionally evaluated by cultivation-based methods of sputum and bronchoalveolar lavage (BAL); the magnitude and type of organisms have varied widely.69 72 Loose correlations have been reported between the identification of bacteria and the intensity of the inflammatory/immune response. Similarly, bacterial colonisation detected with these techniques has been associated with increased cough and sputum71 and increased rates of AECOPD.71–73 Varying bacterial strains in sputum have been linked with increased likelihood of AECOPD.74 A key limitation of cultivation-based methodologies lies in their inability to address the issue of unculturable organisms.
Until very recently, modern molecular techniques have been infrequently applied to characterise the lung microbiota in people who smoke or patients with COPD. Bronchoscopic studies indicate that the lungs of healthy people who smoke are inhabited by diverse types of bacteria in relatively small numbers and that this microbiome changes with disease.1 75 In five patients with COPD with moderate spirometric disease and eight controls, bronchoscopic and BAL defined microbiota were similar.1 In eight mechanically ventilated patients with AECOPD, a diverse microbial community was identified, despite recent or ongoing antibiotic exposure.75 In a separate study of BAL in seven people who smoke with normal lung function, four patients with COPD, and three healthy controls,2 surprisingly significant levels of bacterial 16S gene signal were detected in all three study groups with no significant differences between the groups in the number of bacterial copies (figure 2). The microbiome of patients with moderate or severe COPD was notable for decreased bacterial community diversity, although there was extensive overlap in membership among the bacterial communities of the three groups. These data also suggest that there may be bacteria that comprise a ‘core’ pulmonary microbiome, that is, found with very high frequency (at >1% of all 16S reads) in the BAL of healthy controls. Candidate genera that were found in >75% of these controls included Pseudomonas, Streptococcus, Prevotella and Fusobacteria. Haemophilus, Veillonella and Porphyromonas were also identified in over half of the samples.

Taxonomic classification of bronchoalveolar lavage samples of healthy people who smoke, patients with chronic obstructive pulmonary disease (COPD) and people who do not smoke with normal spirometry at a phylum level.
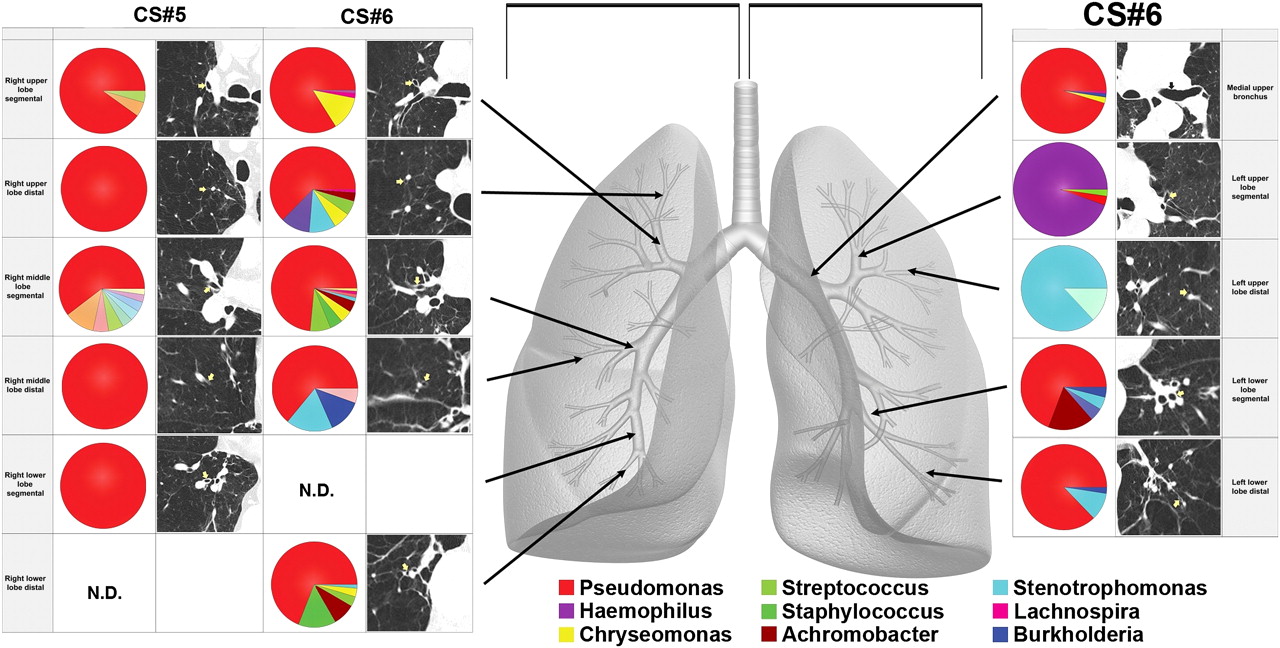
In that same study, the microbiome was also examined in explants removed at lung transplantation in six patients with severe COPD (mean forced expiratory volume in 1 s 16% of predicted); significant differences in bacterial community composition within the same lung were seen (figure 3). Ordination by principal component analysis demonstrated that the micro-anatomic variation in the samples was driven by the dominance of Pseudomonas, Haemophilus or Stentrophomonas at the site. Analysis of OTUs (based on 3% dissimilarity) demonstrated that there were significant numeric differences in the number of OTUs in tissue sites within the same lung and even within the same lobe. Hence marked micro-anatomic differences can exist in bacterial communities even within the same lung of patients with advanced COPD. These findings may have an important biological impact on disease pathogenesis.
{kind=link}
{kind=link}
{kind=link}
Regional heterogeneity is noted in lung microbiota. Multiple samples were harvested from the regions of lung indicated by the arrows on the grey lung schematic in two chronic obstructive pulmonary disease (COPD) explants. Pie diagrams depict the genus level classification of 16S sequences, and the CT images demonstrate the absence of bronchiectasis in the airways adjacent to where samples were obtained. The key for the nine most abundant organisms is provided below the lung schematic. Significant heterogeneity is seen within the lungs from these patients with COPD. ND, not done.
Microbiome in CF
Chronic infection and inflammation of the airways remain the leading causes of morbidity and mortality in patients with CF. Traditional culture-based microbiology has long shown that the airways of most patients with CF become infected, in a fashion that is largely temporally stereotypic, with a relatively small set of bacterial species, including Staphylococcus aureus, H influenzae and Pseudomonas aeruginosa. Other opportunistic species, such as Burkholderia cepacia complex, Stenotrophomonas maltophilia and Achromobacter xylosoxidans, are less frequently recovered in cultures from older patients.76
Recently, culture-independent approaches have led to a greater appreciation of the complexity of CF airway infection. Collectively, analyses that rely on extracting phylogenetic information from the bacterial 16S rRNA gene through Sanger sequencing of clone libraries,77 78 T-RFLP analysis,79–81 microarray hybridisation82 83 or pyrosequencing84 85 to assess the microbiota of the CF airways have revealed the presence of far more diverse bacterial communities than had been appreciated with culture-based methods. The range of bacterial species identified in CF airways has increased from a handful of easily cultured species to dozens of taxa, including several species of facultative and obligate anaerobic bacteria (eg, Prevotella, Veillonella, Propionibacterium spp. and Streptococcus milleri) and aerobes rarely recovered in respiratory cultures (eg, Granulicatella, Gemella and Rothia spp.). Many of these species are regarded as members of the oropharyngeal flora. As such, it remains to be determined whether true infection exists or whether their presence reflects defective clearance of aspirated organisms. These possibilities may not be mutually exclusive and the latter may reflect a legitimate insult to the innate and adaptive immune system that contributes to disease pathogenesis. Regardless of the source of these complex microbial assemblages, studies with the CF pig model suggest that complex polymicrobial infection of the CF airways may begin quite early in infancy or childhood, prior to demonstrable clinical or radiological signs of infection.86
Recent studies have also indicated that microbial communities in CF are dynamic. In one study including a wide age range of children and adults with CF, bacterial diversity and increased phylogenetic relatedness of bacterial communities were associated with increased age and decreased lung function.82 Klepac-Ceraj and colleagues83 reported similar findings in a population of paediatric patients with CF. Bacterial community dynamics may also vary by CFTR genotype.82 A limitation of these studies is that they are largely cross-sectional, with serial samples available from only a small number of patients followed longitudinally. In a study of six adult patients with CF, which performed pyrosequencing analysis of 126 sputum samples serially recovered over 10 years, decreases in bacterial community diversity (measured by Shannon Index) were not invariably associated with advancing age, depending on patient CFTR genotype (J LiPuma, unpublished data). Again, decreased diversity was associated with disease severity and antibiotic use; however, determining the causal relationship between these confounding variables presents a significant challenge. This difficulty is compounded by intensive and extremely variable antibiotic use among patients with CF, particularly those with advanced lung disease. Although decreased community diversity appears to correlate in general with long-term antibiotic use, the short-term use of antibiotics seems to represent a relatively minor disturbance.87 Antibiotic treatment of exacerbation results in reduced density of many species; however, the effect appears to be largely transient, and bacterial density as well as diversity is rapidly restored (Wolfgang & Boucher, Kong and Lynch, unpublished data). It remains to be determined whether this degree of resiliency in community structure is maintained during the advanced stages of lung disease. The availability of culture-independent methods to study the microbiota of CF airways presents an extraordinary opportunity to advance our understanding of what is increasingly recognised as a very complex microbial ecology.
Future directions
Genomic microbial analysis of the lung is a relatively young field. Basic studies identifying the bacterial composition of the healthy lung and the relationship between the upper and lower airway microbiome in both healthy people and those with specific lung diseases are still needed. Although the lungs were not included in the original Human Microbiome Project, these questions are currently being addressed in the USA by several large trials sponsored by the NHLBI.88 89 A better understanding of the relationship between findings from culture-dependent and molecular microbiological techniques is also needed, as the current literature yields conflicting results.1 2 28 64 75 The ultimate goals of this research area are to understand whether specific aspects of the lung microbiome relate to disease progression and whether manipulating the bacterial community might preserve lung function. Because asthma, COPD and CF are all characterised by periodic exacerbations, other important areas of future research are to define how bacterial communities change around the time of exacerbation, and even whether changes in the bacterial community structure might predict exacerbation, possibly allowing early or preemptive intervention table 1.
References
Footnotes
Dr Toews died on 12th Oct 2011.
Funding MKH is supported by NHLBI grant K23 HL093351. YJH is supported by NIH/NHLBI grant 1K23HL105572-01 and a Cystic Fibrosis Foundation PACE award. JJL is supported by NIH/NHLBI grant 1RC1HL100809-01 and CTSA grant UL1RR024986 and the Cystic Fibrosis Foundation. JLC is supported by NIH/NHLBI grant R01 HL082480 and by a Research Enhancement Award Program from the Biomedical Laboratory Research & Development Service, Department of Veterans Affairs. GBH is supported by grants RO1-AI064479, R21-AI087869, R21-AI083473, R21-AI087869 and P30-DK034933. FJM is supported by NIH/NHLBI contracts HHSN2682011008C and HHSN268200900016C. SVL is supported by grants AI075410, U01HL098964, AI113916, R21AT004732, PBBR, CFF and CFRI awards. VBY is supported by grants UH3 DK083993, U19 AI090871, U01 HL098961, R01 DK070875 and P30 DK034933.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.