Article Text

Abstract
Background Systemic blockade of tissue factor (TF) attenuates acute lung injury (ALI) in animal models of sepsis but the effects of global TF deficiency are unknown. We used mice with complete knockout of mouse TF and low levels (∼1%) of human TF (LTF mice) to test the hypothesis that global TF deficiency attenuates lung inflammation in direct lung injury.
Methods LTF mice were treated with 10 μg of lipopolysaccharide (LPS) or vehicle administered by direct intratracheal injection and studied at 24 h.
Results Contrary to our hypothesis, LTF mice had increased lung inflammation and injury as measured by bronchoalveolar lavage cell count (3.4×105 wild-type (WT) LPS vs 3.3×105 LTF LPS, p=0.947) and protein (493 μg/ml WT LPS vs 1014 μg/ml LTF LPS, p=0.006), proinflammatory cytokines (TNF-α, IL-10, IL-12, p<0.035 WT LPS vs LTF LPS) and histology compared with WT mice. LTF mice also had increased haemorrhage and free haemoglobin in the airspace accompanied by increased oxidant stress as measured by lipid peroxidation products (F2 isoprostanes and isofurans).
Conclusions These findings indicate that global TF deficiency does not confer protection in a direct lung injury model. Rather, TF deficiency causes increased intra-alveolar haemorrhage following LPS leading to increased lipid peroxidation. Strategies to globally inhibit TF may be deleterious in patients with ALI.
- Airway Epithelium
- ARDS
Statistics from Altmetric.com
Introduction
Activation of the extrinsic coagulation cascade through upregulation of tissue factor (TF)-dependent procoagulant activity has been implicated in the pathogenesis of acute lung injury (ALI) and contributes to lung inflammation. In the setting of ALI, TF protein is elevated systemically1 and locally2 within the airspaces. Systemic blockade of TF in sepsis attenuates organ dysfunction and ALI. He et al3 showed that systemic blockade of TF in an intestinal ischaemia-reperfusion model attenuated the severity of lung injury, leak and inflammation. Welty-Wolf and colleagues found that systemic blockade of TF using either TF blocking antibody4 or active site inactivated factor VIIa5 ,6 attenuated lung injury in an Eschericia coli sepsis model in baboons. This group further reported that systemic blockade of TF activity attenuated lung inflammation in a model of direct lung injury using intratracheal lipopolysaccharide (LPS).7 In summary, there is ample evidence that systemic inhibition of TF attenuates lung inflammation and injury induced by direct and indirect insults. Despite this, the mechanisms by which blockade of TF activity modulates coagulant and inflammatory processes in the lung are not clear. Studies by our group and others have shown that intra-alveolar fibrin deposition is modulated locally within the airspace by resident lung cells including the lung epithelium,2 ,8–10 but effects of TF inhibition on lung coagulation remain incompletely understood.
Given the compelling evidence that systemic blockade of TF ameliorates lung injury in indirect injury (sepsis) models but the paucity of data on the effects of global TF inhibition on acute lung inflammation caused by direct lung injury, we designed a series of experiments to test the hypothesis that global TF deficiency is protective in a model of direct lung inflammation. We used genetically manipulated mice that have global absence of murine TF (mTF) but express human TF protein at 1% of endogenous levels to prevent embryonic lethality (LTF mice).11 Importantly, these mice have a haemostatic defect and exhibit spontaneous haemorrhage in various tissues, including the lung.12 Histological analysis of lungs from 6-month old LTF mice showed extensive hemosiderin deposition suggestive of chronic lung haemorrhage.12 Despite this, LTF mice were protected in an indirect lung injury (endotoxemia) model13 with increased survival following systemic administration of endotoxin. How genetic deficiency of TF affects lung-specific coagulation and inflammation is unknown. Here we present the results of experiments using a model of direct lung injury, intratracheal LPS, in LTF mice and wild-type (WT) littermate controls.
Materials and methods
Transgenic mice
The Vanderbilt Institute for Animal Care and Use Committee approved the study protocol. Transgenic LTF mice on a C57/BL6 background were generated as previously described by Parry et al11 and expressed human TF mRNA at ∼1% of levels measured in WT mice. LTF mice (mTF−/−, hTF+, N=51) and WT littermate controls (mTF+/+, hTF−, N=38) were compared.
Mouse experimental protocol and tissue collection
Mice were anesthetised with isoflurane and instilled by direct intratracheal injection14 with 100 μl of 100 μg/ml LPS (E coli, LPS; Sigma, St Louis, Missouri, USA), 100 μl of phosphate-buffered saline (PBS; control) (Mediatech, Manassas, Virginia, USA) and with 100 nM recombinant mTF (R&D Systems, Minneapolis, Minnesota, USA) for selected experiments. All data represent 2–12 mice per group as noted. Analysis was done using all available samples for each measurement. Limited sample volumes did not allow every measurement to be made in every animal. BAL was obtained by instilling 900 μl 0.9% NaCl and aspirating the fluid. BAL was centrifuged at 1000 g for 10 min and supernatant was frozen at −80°C. The lungs were removed, flash frozen in liquid nitrogen and stored at −80°C.
Bronchoalveolar lavage cell count and differential
Manual cell counts (hemocytometer) and cytospins/differentials were completed with fresh bronchoalveolar lavage (BAL) fluid. Cytospins were stained with Hema 3 staining kit (Thermo-Fisher Scientific, Pittsburg, PA, USA).
Lung mTF Western
Protein was extracted from frozen lung tissue using radioimmunoprecipitation assay buffer containing 50 mM Tris, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.5% sodium deoxycholate, 1% Triton-x-100 and 0.1% sodium dodecyl sulphate (SDS) with Protease Inhibitor Cocktail (Roche, Indianapolis, Indiana, USA) and kept on ice using standard methodology. Protein was separated by SDS polyacrylamide gel electrophoresis, using 10% polyacrylamide gels (BioRad, Hercules, California, USA), electro blotted onto a polyvinylidene difluoride membrane (Millipore, Burlington, Massachusetts, USA) and blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, New Brunswick, Canada), washed with PBS 0.1% Tween and incubated with anti-TF primary antibody (R&D Systems) at1 : 1000 overnight at 4°C. Blots were washed and incubated with secondary antibody (LI-COR) at 1 : 15 000 for 1 h at room temperature and densitometric measurements were made with the Odyssey infrared imaging system (LI-COR) with actin as a protein loading control.
Lung hTF PCR
RNA was isolated from 30 mg of lung tissue with the Pure Link RNA Mini Kit (Invitrogen, Grand Island, NY, USA) with Trizol (Invitrogen). cDNA was synthesised using SuperScript VILO kit (Invitrogen). Real-time PCR assay was performed using Taqman Universal Master Mix and commercially available primers/probes for human TF (Applied Biosystems, Carlsbad, California, USA) on a Step-One PCR instrument (Applied Biosystems).
mTF immunostaining
Paraffin-embedded lung sections were de-paraffinised with antigen retrieval by standard procedures. Slides were incubated overnight at 4°C with 1 : 200 goat polyclonal anti-TF Ab (R&D Systems), rinsed and incubated with a biotinylated rabbit anti-goat secondary antibody (Innogenex, San Ramon, California, USA) for 20 min at room temperature. Slides were developed with NovoRed (Vector, Burlingame, California, USA) for 30 s and counterstained with methyl green for 10 s.
Lung wet-to-dry weight ratio
The left lung was placed on pre-weighed aluminium foil, weighed and placed in an 80°C oven for 2 days. The foil+lung was weighed again and the ratio was calculated ((lung−foil before drying)/(lung−foil after drying)).
BAL clot time measurements
Clot time was measured using a mechanical clot detection system (STart4 Coagulometer; Diagnostica Stago, Paris, France). Twenty-five microlitres of BAL was warmed for 15 min at 37°C then incubated with 25 μl of pooled citrated mouse plasma (Bioreclamation, East Meadow, New York, USA). Clot time was determined in duplicate as recalcification time following the addition of 25 μl of 50 mM calcium chloride.
BAL protein and cytokine measurements
Protein was measured in BAL using the BCA protein assay (Pierce, Rockford, Illinois, USA). Cytokines were measured by electrochemiluminescence in a mouse 7-plex proinflammatory cytokine assay (Meso Scale Discovery, Gaithersburg, Maryland, USA).
Lung histology and scoring
Formalin-fixed and paraffin-embedded lung sections were stained with H&E. Ten images were captured at 40× power across two lobes of each lung. Each image was scored on a five-point scale modified from Frank et al15 for septal thickening, oedema, inflammation and haemorrhage in a blinded fashion. The scores for each parameter were averaged for the 10 images and the averages added to generate a total lung injury score.
Free haemoglobin measurements
Free haemoglobin was measured in undiluted cell-free pulmonary oedema fluid collected as previously described2 from critically ill mechanically ventilated patients with ALI or control patients with severe hydrostatic pulmonary oedema (HYDRO) using the low haemoglobin analyser from HemoCue (Cypress, California, USA). Human studies were approved by the Vanderbilt Institutional Review Board.
Lung isoprostane and isofuran measurements
F2 isoprostanes and isofurans were quantified in mouse lung homogenates and human BAL by stable isotope dilution gas chromatography/negative ion chemical ionisation mass spectrometry as described previously.16 ,17
Statistical analysis
Non-normally distributed data were natural log transformed prior to analysis and are displayed as boxplots or dot plots. Normally distributed data are displayed as bar graphs showing mean and SEM. Comparison of three or more groups was done using one-way analysis of variance with post hoc Tukey test. Comparison of the two groups was by Student's t test except for percent weight loss which was non-normally distributed but could not be log transformed because of negative numbers. For this analysis, a Kruskall–Wallis test was followed by pairwise Mann–Whitney U tests for comparisons between groups. All analyses were done with SPSS software V.19 for Macintosh. p<0.05 was considered statistically significant.
Results
TF levels and coagulation in LTF mice
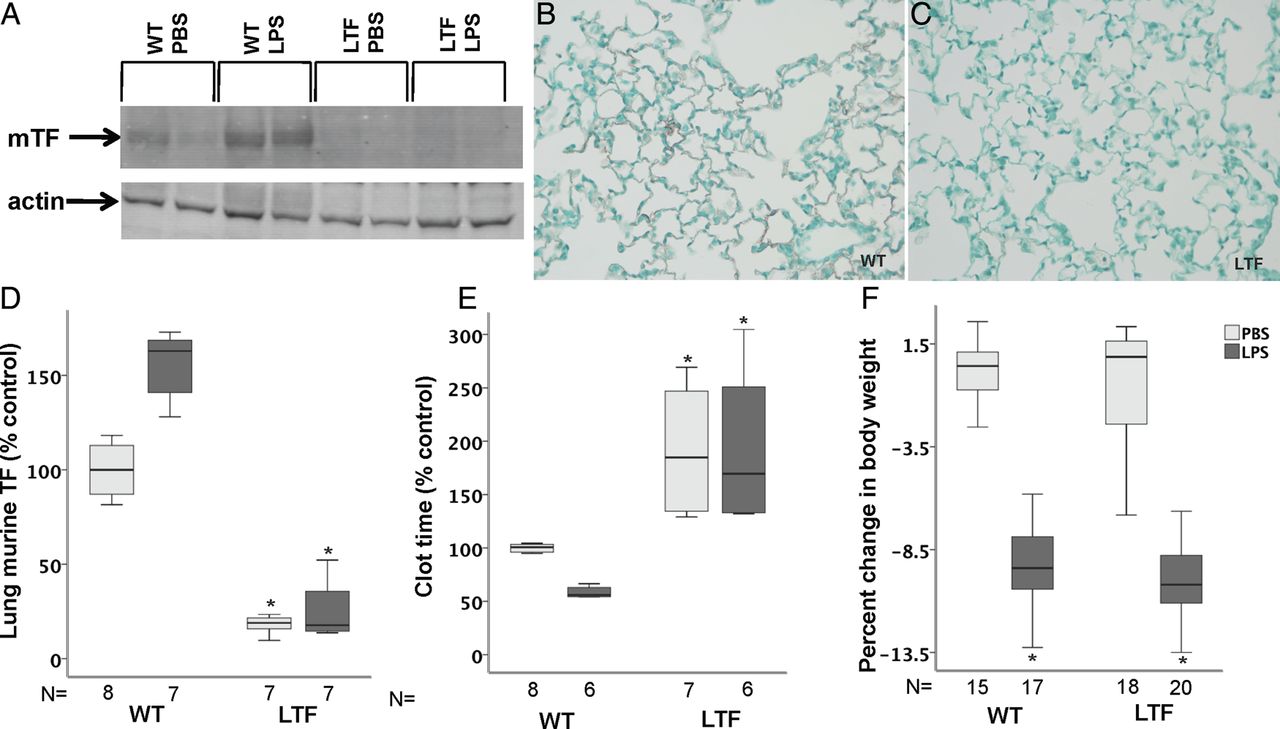
mTF was present in low levels in lung homogenates in WT mice and modestly increased following treatment with intratracheal LPS (p=0.116 WT PBS vs WT LPS) (figure 1A and D). LTF mice had no detectable mTF in either treatment group. Immunostaining showed mTF protein lining the airspace in WT mice (figure 1B) but no detectable staining in LTF mice (figure 1C). hTF mRNA was detectable in LTF but not in WT mice and LPS treatment had no effect on hTF expression (not shown). In WT mice, clot time decreased with LPS compared with PBS treatment (figure 1E) consistent with induction of TF-dependent procoagulant activity in the airspace. In LTF mice, BAL clot time was prolonged compared with WT mice (figure 1E) in both treatment groups consistent with the deficiency of TF and was not significantly shortened in response to LPS. WT and LTF mice lost significant body weight 24 h after treatment with intratracheal LPS (figure 1F).
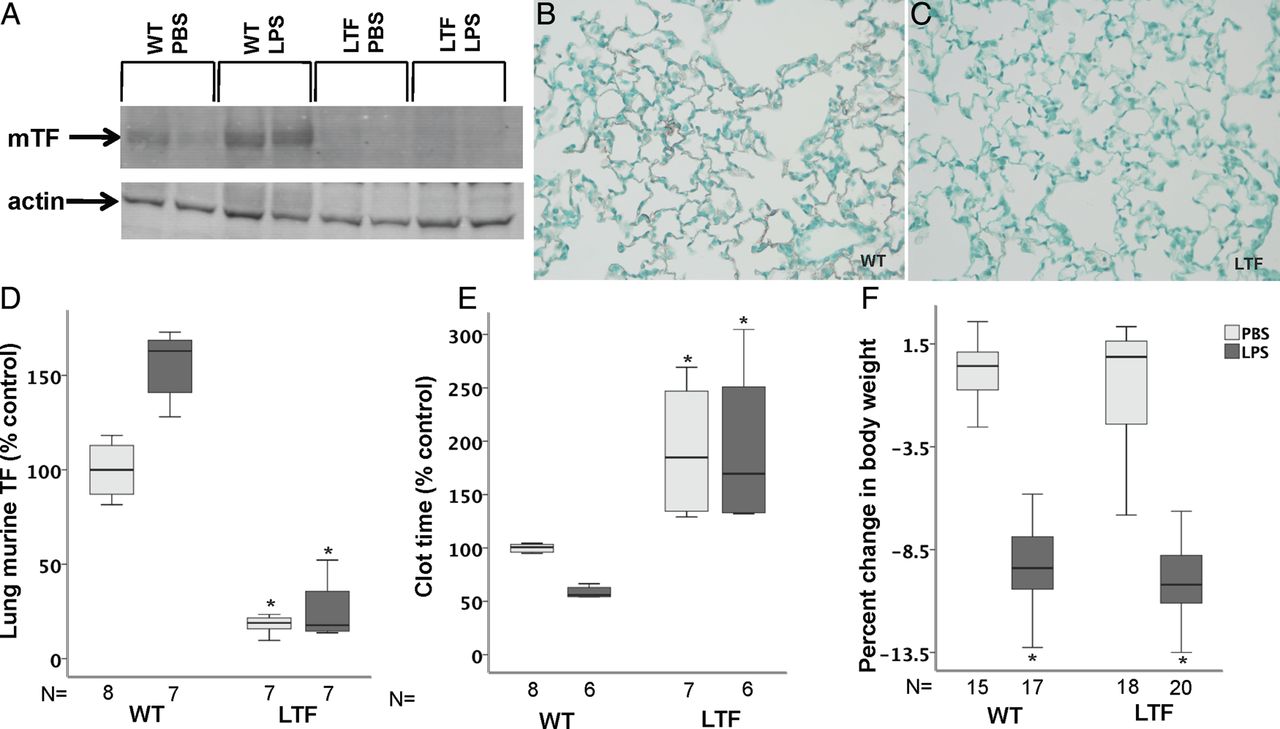
Tissue factor (TF) expression. Wild-type (WT) mice have low but detectable levels of murine TF (mTF) expression at baseline that increases following intratracheal lipopolysaccharide (LPS) treatment while mTF levels are undetectable in low TF (LTF) mice. (A) A representative western blot for mTF and (D) densitometric quantification from four independent experiments. *p=<0.001 versus both WT groups. Immunostaining for mTF (red/brown) in WT (B) and LTF (C) mice 24 h after intratracheal LPS. While WT mice have a shortened bronchoalveolar lavage clot time in response to intratracheal LPS (E), LTF mice have a prolonged clot time that is not shortened by intratracheal LPS. *p<0.001 versus WT. (F) Weight change 24 h after treatment with LPS. *p<0.001 versus phosphate-buffered saline (PBS) treatment.
LTF mice are not protected from lung inflammation induced by intratracheal LPS
WT and LTF mice had increased BAL total cell counts 24 h after intratracheal LPS (figure 2A). One-third of the LTF mice had BAL cell counts that were higher than the highest counts in WT mice. In WT mice treated with LPS, neutrophils predominated (88% neutrophils, 12% macrophages) while LTF mice had a higher percentage of macrophages (45% neutrophils, p=0.513 vs WT LPS, 55% macrophages, p=0.513 vs WT LPS) (figure 2B). Additionally, LPS-treated LTF mice had significantly increased BAL levels of interleukin (IL)-10, IL-12 and tumour necrosis factor α and a non-significant increase in interferon γ, IL-1β, IL-6 and IL-8 (table 1).
Median (IQR) BAL cytokines in WT and LTF mice treated with PBS or LPS

Intratracheal lipopolysaccharide (LPS) induces lung inflammation. Dot plot of bronchoalveolar lavage (BAL) cell counts 24 h after intratracheal LPS administration. BAL total cell count was increased in wild-type (WT) and low tissue factor (LTF) mice compared with saline-treated animals (A). *p<0.005 versus phosphate-buffered saline (PBS) treatment by analysis of variance of log-transformed data with post hoc Tukey test. (B) Median (IQR) total BAL macrophage and neutrophil counts and percent counts.
There was also a modest increase in IL-6 and IL-8 in PBS-treated LTF mice compared with WT.
LTF mice have increased pulmonary oedema in response to intratracheal LPS
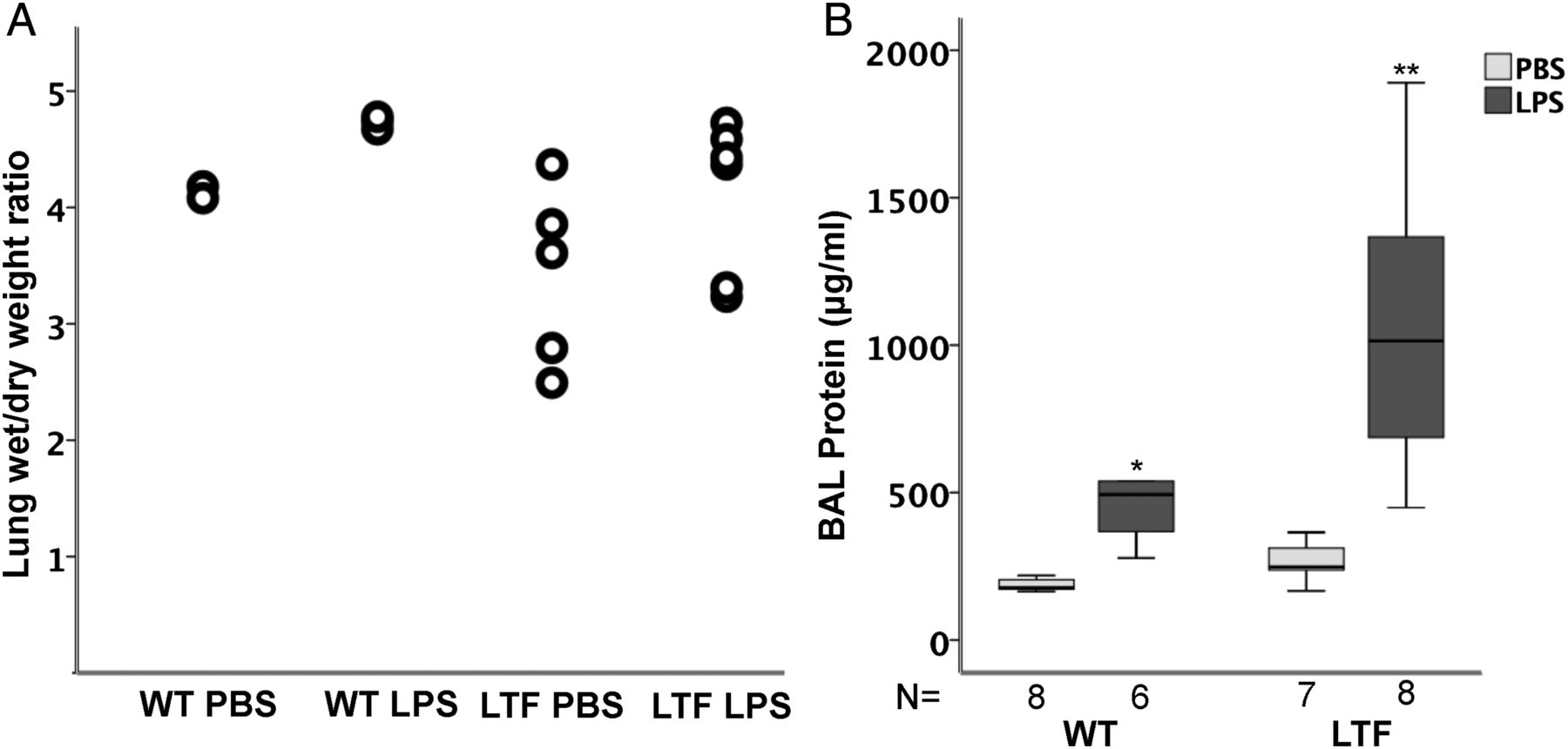
WT and LTF mice had modest but non-significant increases in wet-to-dry weight ratio (figure 3A) following treatment with intratracheal LPS without a significant difference between genotypes. BAL protein, however, was increased in response to intratracheal LPS in both groups (figure 3B) and LTF mice had BAL protein levels that were twofold higher than WT mice.
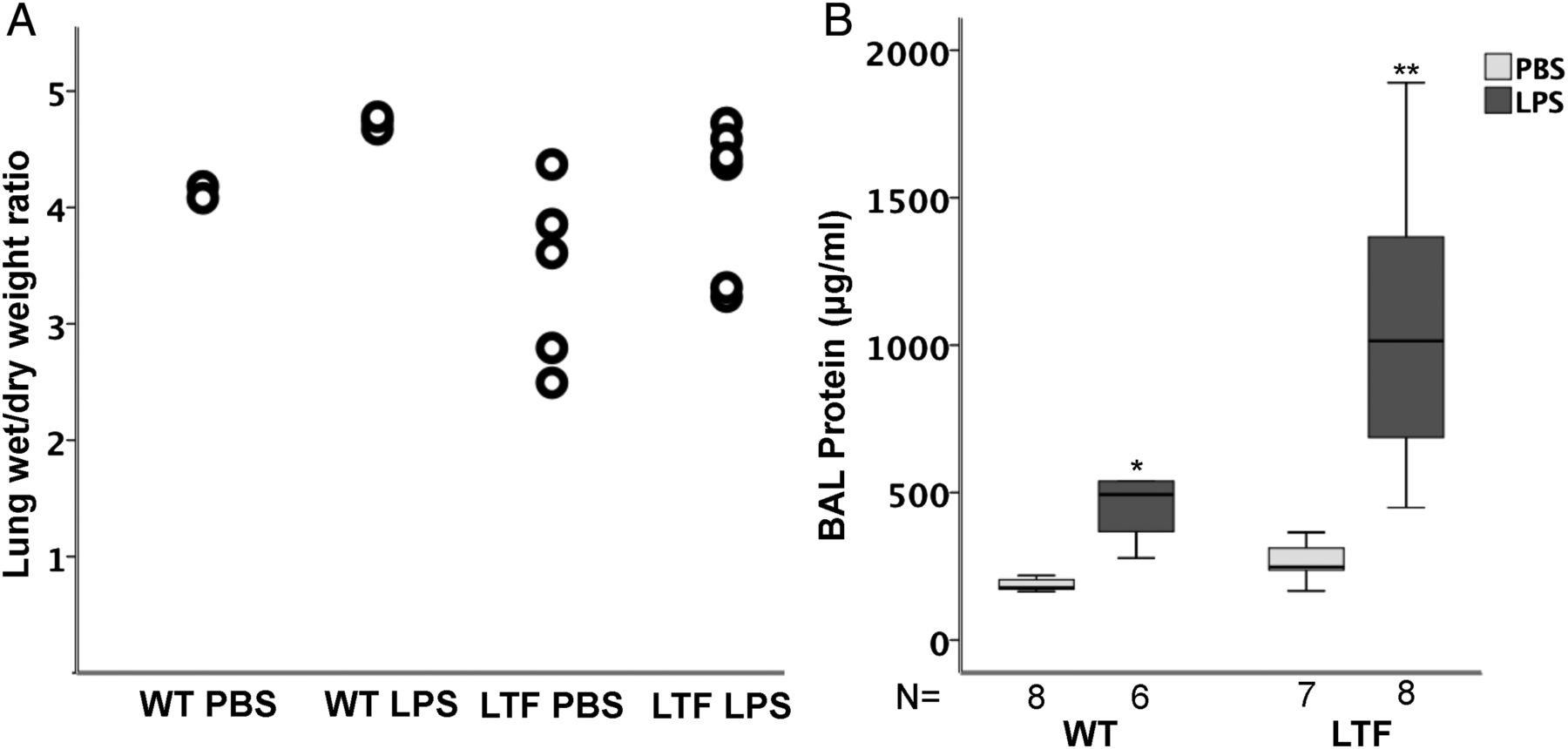
Intratracheal lipopolysaccharide (LPS) increases lung oedema. Intratracheal LPS causes a modest but non-significant increase in lung wet to dry weight ratio (A) but there are no differences between wild-type (WT) and low tissue factor (LTF) mice. In contrast, bronchoalveolar lavage (BAL) total protein is significantly increased by intratracheal LPS (B). *p<0.007 versus WT PBS and LTF LPS groups, **p<0.007 versus all other groups.
WT and LTF LPS-treated mice have increased histological lung injury
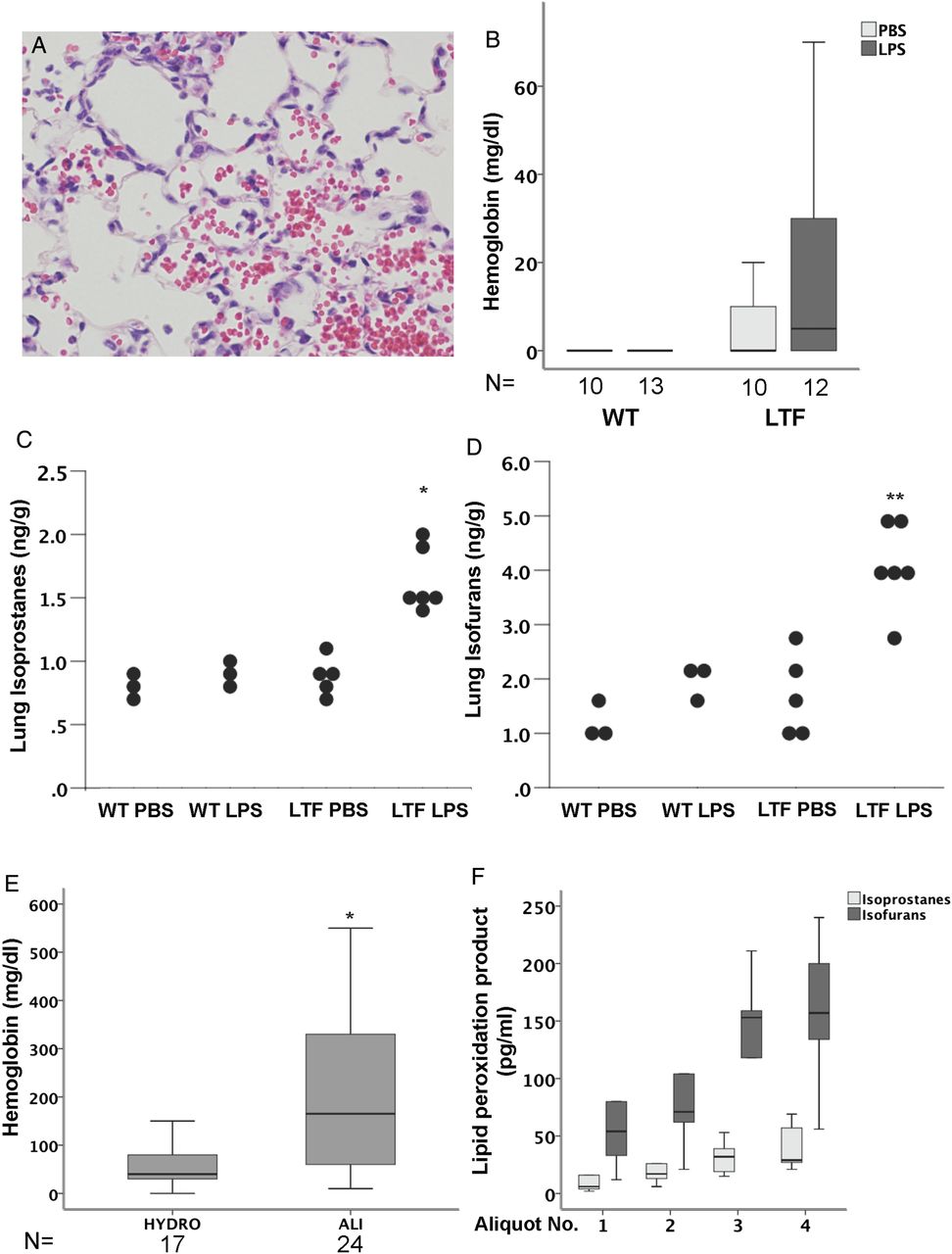
WT (figure 4A) and LTF (figure 4B) mice had increased histological evidence of lung injury 24 h after intratracheal LPS (figure 4C, E). LTF mice had significantly more alveolar haemorrhage (figure 4D,E, figure 5A) compared with WT mice.
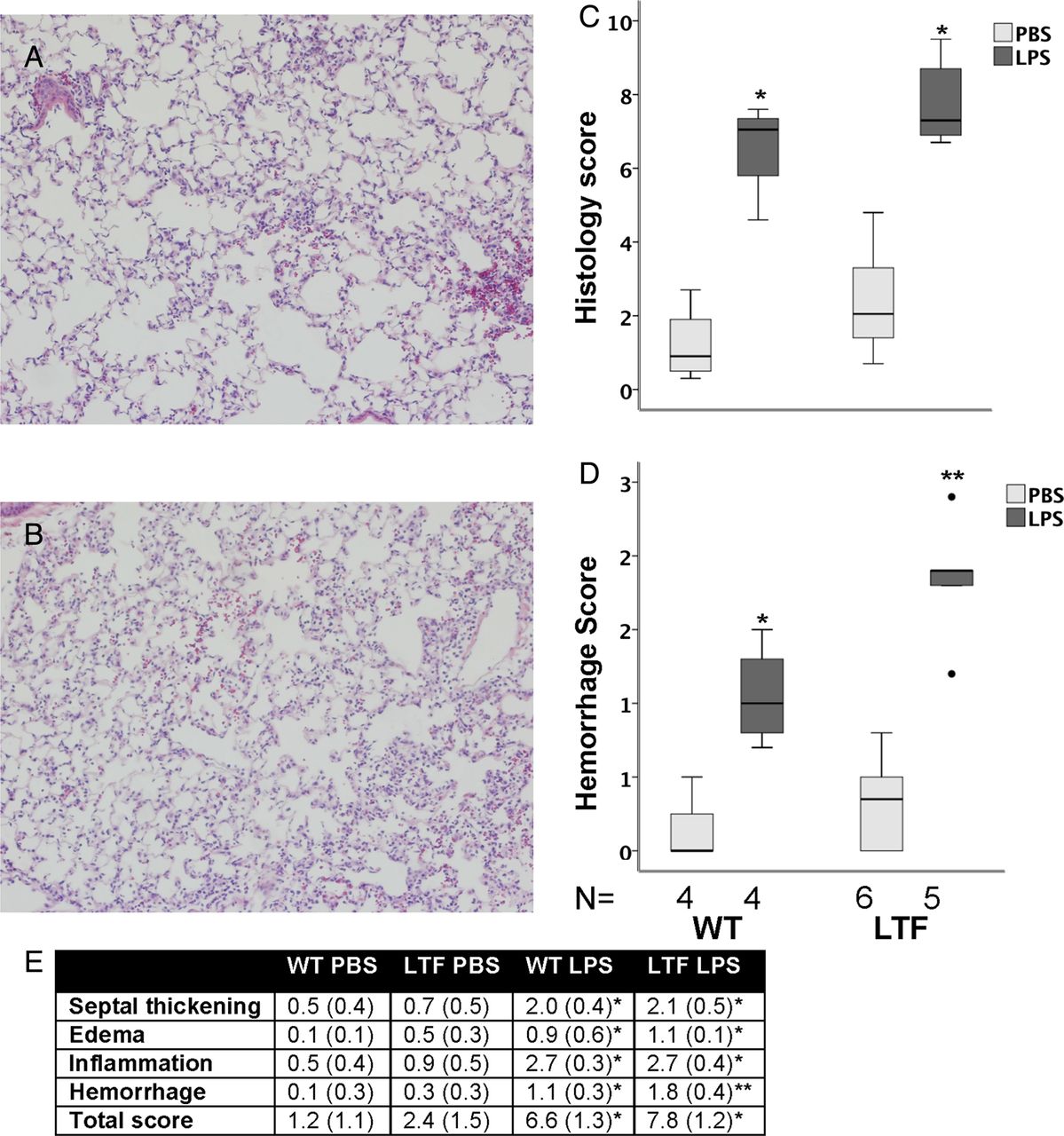
Lung histology 24 h after intratracheal lipopolysaccharide (LPS). Intratracheal LPS-induced lung inflammation and injury in wild-type (WT) (A) and low tissue factor (LTF) (B) mice. LPS treatment increased lung injury in both genotypes (C) (*p<0.002 vs phosphate-buffered saline (PBS) treatment). Lung haemorrhage was more severe in LPS-treated LTF mice compared with WT mice (D) (*p<0.027 vs all other groups; **p<0.019 vs all other groups). (E) Individual components of the score (*p<0.05 vs PBS treatment in both genotypes; **p<0.05 vs PBS treatment in both genotypes and WT LPS treatment).

Free haemoglobin and lipid peroxidation. Low tissue factor (LTF) mice have increased alveolar haemorrhage (A) and increased free haemoglobin in bronchoalveolar lavage (BAL) fluid (B) basally and in response to intratracheal lipopolysaccharide (LPS). LTF mice also have an increase in total lung isoprostanes (C) and isofurans (D) in response to intratracheal LPS (*p<0.001 vs all other groups and **p<0.021 vs all other groups). Patients with attenuates acute lung injury also have increased levels of free haemoglobin in the lung compared with patients with hydrostatic oedema (E) (*p=0.007). In patients with a diagnosis of alveolar haemorrhage, BAL isoprostanes and isofurans increase with increasing aliquot number (F).
LTF mice have higher levels of free haemoglobin, isoprostanes and isofurans in the lung compared with WT mice
Because LTF mice were not protected from intratracheal LPS-induced lung inflammation and had increased intra-alveolar bleeding, we hypothesised that release of free haemoglobin into the airspaces might be contributing to lung injury and inflammation in the LTF mice. LTF mice treated with intratracheal LPS had higher levels of free haemoglobin in the cell-free BAL than WT mice (figure 5B). To test whether the free haemoglobin is contributing to lung injury through oxidant stress we measured isoprostanes (figure 5C) and isofurans (figure 5D) in lung homogenates. LPS-treated LTF mice had higher levels of both markers in the lung compared with LPS-treated WT mice. To test whether these findings are applicable to human disease, we measured free haemoglobin in undiluted pulmonary oedema fluid from patients with ALI compared with control patients with hydrostatic pulmonary oedema. Free haemoglobin levels were significantly elevated in patients with ALI (figure 5E). Finally, to test whether free haemoglobin in human lungs is associated with markers of lipid peroxidation we measured BAL isoprostanes and isofurans in sequential BAL aliquots from five patients with diffuse alveolar haemorrhage (figure 5F) in whom sequential aliquots of BAL fluid were progressively bloodier. As the aliquot number increased, BAL isoprostane and isofuran levels increased.
Effects of mTF replacement in LTF mice
LTF mice were treated with LPS or LPS plus recombinant mTF and analysed at 24 h. LTF mice treated with mTF had visibly decreased BAL haemorrhage (figure 6A) and a shortened clot time (figure 6B) suggesting that replacement of TF into the airspace partially restored TF procoagulant activity. mTF treatment attenuated lung permeability induced by intratracheal LPS as measured by BAL protein (figure 6C) but did not affect inflammatory cell influx (figure 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of tissue factor (TF) replacement in low TF (LTF) mice. LTF mice were treated with 10 μg intratracheal lipopolysaccharide (LPS) (N=4) or 10 μg lipopolysaccharide (LPS) plus 100 nM intratracheal recombinant murine TF (mTF) (N=5) and harvested at 24 h. With the administration of mTF, LTF mice had visibly reduced BAL haemorrhage (A) and significantly shortened BAL clot time (B) compared with LPS-treated mice that did not receive recombinant TF. Bronchoalveolar lavage (BAL) total protein was higher in mice given mTF (C) but BAL cell counts did not differ (D) (*p=0.011; **p=0.031).
Discussion
Contrary to our hypothesis, global deficiency of TF is not protective in a model of direct lung inflammation. LTF mice have influx of inflammatory cells into the lung in response to intratracheal LPS with BAL cell counts that are comparable to WT mice. In addition, LTF mice have increased BAL cytokines, histological severity of lung injury and alveolar capillary barrier permeability compared with WT mice. These findings are in distinct contrast to prior reports that systemic inhibition of TF was protective in either a direct or indirect model of ALI.4–7 Interestingly, the same LTF mice treated with systemic LPS have decreased systemic inflammation and improved survival compared with control mice.18 While the increases in lung injury in LTF mice in our studies are modest, the fact that our findings contradict prior work in this field is particularly interesting.
TF-mediated coagulation is upregulated in the lung in the setting of inflammation and injury2 ,19–21 but whether TF in the lung is protective or injurious is not completely understood. Others have shown that TF inhibition and deficiency protects against lung injury. One important difference between our study and others is that we used a model of global TF deficiency while the previous studies administered pharmacological TF inhibitors systemically, which may or may not have modulated coagulation in the lung. We have previously shown that coagulation is modulated locally by the lung epithelium.2 ,8 ,9 ,22 Because of this local regulation, modulation of coagulation systemically in the previous studies might have very different effects compared with global TF deficiency. Our findings suggest that mice with global TF deficiency respond quite differently to local versus systemic inflammatory insults.
An early observation in our studies was that BAL from LTF mice appeared blood tinged while the BAL from WT mice was clear. LTF mice are known to have chronic haemorrhage into the lungs12 and might be expected to have increased haemorrhage in response to injury. In addition, LTF mice have increased lung permeability which may contribute to increased bleeding in the lungs. The discrepancy between BAL protein and wet-to-dry weight ratios may be explained by the relatively mild epithelial injury in LTF mice that allows for preserved alveolar fluid clearance in the setting of increased permeability. To understand whether blood in the lung augmented lung inflammation, we studied the potential injurious effects of bleeding into the lungs. In addition to intact red blood cells in the airspace as seen on histology, LTF mice had red-tinged BAL fluid after the removal of cellular components, suggestive of free haemoglobin. Only 13% of WT mice had any detectable free haemoglobin in the BAL after LPS while 41% of LTF mice had measureable levels (figure 5B). Since free haemoglobin is a potent oxidant and could potentially contribute to lung inflammation and injury, we measured lung isoprostanes and isofurans as markers of oxidant injury and found that both were elevated in the lungs of LTF mice compared with WT mice (figure 5C,D). Thus, one potential mechanism whereby LTF mice have increased lung inflammation and injury is through the harmful effects of free haemoglobin, a potent inducer of oxidant stress. Free haemoglobin oxidises lipids and can cause extensive cell damage.23 Haemoglobin can potentiate vascular injury induced by oxidised LDL24 or activated neutrophils.25 Although cell-free haemoglobin has been implicated in the pathogenesis of transfusion related ALI,26 ,27 little is known about the direct effects of free haemoglobin in the airspace. Our data suggest that free haemoglobin in the airspace may be injurious by inducing lipid peroxidation.
To determine the clinical significance of our findings, we measured free haemoglobin or isoprostanes/isofurans in the airspaces of two patient cohorts. Patients with ALI had higher levels of free haemoglobin in the airspace compared with patients with hydrostatic pulmonary oedema. Patients with diffuse alveolar haemorrhage had progressively increasing BAL isoprostanes and isofurans as their sequential BAL aliquots become more bloody. These findings suggest that liberation of free haemoglobin into the airspace and intra-alveolar lipid peroxidation may be important mechanisms of clinical ALI.
To confirm that the effects of LPS in LTF mice were a result of TF deficiency, we reconstituted lung TF by coinjection of recombinant mTF with LPS. mTF replacement attenuated lung haemorrhage and permeability and augmented lung coagulation but had no effects on lung inflammation. These data suggest that free haemoglobin mediates the increased lung permeability and lipid peroxidation in LTF mice and can be reversed by TF replacement while TF replacement does not mediate changes in inflammation. Others have shown that circulating free haemoglobin mediates vascular permeability. In a murine model of sickle cell disease, free haemoglobin induces lung endothelial permeability.28 Our study is the first to suggest that free haemoglobin in the airspace may be a potent mediator of lung permeability. Although we have not measured circulating free haemoglobin the fact that replacement of TF in the airspace attenuates lung haemorrhage as well as permeability suggests that it is the intra-alveolar haemoglobin that is mediating these effects. Our study did not specifically test the independent effects of free haemoglobin, so free haemoglobin could also be a marker of increased permeability. In addition to modulating free haemoglobin, the effects of TF deficiency on lung inflammation may be mediated through a different mechanism such as loss of protease-activated receptor (PAR-2) signalling in LTF mice or lack of adequate intra-alveolar TF replacement. Understanding the mechanism of LTF effects on lung inflammation will be the focus of future studies.
The study has some limitations. First, by using mice with a global TF deficiency, we were unable to identify which cell types are most critically involved in the lung response to intratracheal LPS. However, our results do show that global TF deficiency does not protect against direct lung injury, and in fact enhances several indices of ALI. Interestingly, LTF mice may have increased susceptibility to even mild insults. In our LTF mice treated with PBS we observed non-significant increased BAL IL-6 and IL-8, higher levels of BAL protein, and mild increases in histology score and BAL cell counts. This response to a very mild insult highlights the critical nature of TF in limiting lung injury and inflammation. Second, our studies were not designed to test whether the effects of TF deficiency are mediated through the procoagulant or non-coagulant functions of TF, such as signalling effects through PAR-2. Finally, although we have shown that free haemoglobin and markers of lipid peroxidation are elevated in the lungs of LTF mice there may be additional mechanisms that explain the increased lung inflammation seen in these animals.
In summary, mice with a global TF deficiency are not protected from the effects of intratracheal LPS as they have modestly increased lung inflammation, injury and permeability compared with LPS-treated WT mice. Our data suggest that intra-alveolar bleeding in LTF mice in response to intratracheal LPS may contribute to lung injury through the oxidant effects of free haemoglobin. Furthermore, many of the indices of injury can be attenuated with intratracheal TF replacement. These findings are relevant to human ALI and highlight the complexity of coagulation pathways in the acutely injured lung. Two clinical trials related to acute lung inflammation have been completed using recombinant tissue factor pathway inhibitor (TFPI, tifacogin), one in severe sepsis29 and one in severe community-acquired pneumonia.30 Neither of these trials showed a clinical benefit of TFPI, possibly because they included patients with direct and indirect causes of ALI. Furthermore, the specific roles of TF in the lung and vascular compartment may be different as our TF replacement studies might suggest. Future studies will focus on compartmentalised manipulation of the TF pathway in direct and indirect ALI.
Correction notice This article has been corrected since it was published Online First. The author name Jackson L Roberts II has been amended to read L Jackson Roberts II.
Acknowledgments
Acknowledgements The authors would like to thank Bill Zackert for his excellent technical assistance.
References
Footnotes
-
Contributors JAB designed and carried out experiments, analysed data, prepared figures and wrote the manuscript; SCS performed experiments and edited the manuscript; JKC performed experiments and edited the manuscript; BSG performed experiments and assisted with writing the manuscript; WEL helped with experiment design and execution and edited the manuscript; DRJ made the free haemoglobin measurements on the human oedema fluid samples and assisted with editing the manuscript; LJR did the lipid peroxidation measurements and edited the manuscript; RD provided BAL samples from patients with alveolar haemorrhage and assisted with editing the manuscript; NM provided the low TF mice, and assisted with experimental design and manuscript editing; LBW designed experiments, provided human oedema fluid samples, and edited the manuscript.
-
Funding K08 HL 090785 to JAB, T32 HL087738 to SCS, HL 105479 to WEL, NIH GM42056 to LJR, K24 HL103836 and American Heart Association Established Investigator Award to LBW.
-
Competing interests None.
-
Ethics approval Vanderbilt University Institutional Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.