Article Text

Abstract
Background Cough is the most frequent reason for consultation with a family doctor, or with a general or respiratory physician. Treatment options are limited and a recent meta-analysis concluded that over-the-counter remedies are ineffective and there is increasing concern about their use in children. Endogenous inflammatory mediators such as prostaglandin E2 (PGE2) and bradykinin (BK), which are often elevated in respiratory disease states, are also known to cause cough by stimulating airway sensory nerves. However, how this occurs is not understood.
Methods We hypothesised that the transient receptor potential (TRP) channels, TRPA1 and TRPV1, may have a role as ‘common effectors’ of tussive responses to these agents. We have employed a range of in vitro imaging and isolated tissue assays in human, murine and guinea pig tissue and an in vivo cough model to support this hypothesis.
Results Using calcium imaging we demonstrated that PGE2 and BK activated isolated guinea pig sensory ganglia and evoked depolarisation (activation) of vagal sensory nerves, which was inhibited by TRPA1 and TRPV1 blockers (JNJ17203212 and HC-030031). These data were confirmed in vagal sensory nerves from TRPA1 and TRPV1 gene deleted mice. TRPV1 and TRPA1 blockers partially inhibited the tussive response to PGE2 and BK with a complete inhibition obtained in the presence of both antagonists together in a guinea pig conscious cough model.
Conclusion This study identifies TRPA1 and TRPV1 channels as key regulators of tussive responses elicited by endogenous and exogenous agents, making them the most promising targets currently identified in the development of anti-tussive drugs.
- Cough
- sensory nerves
- transient receptor potential channels
- lung
- prostanoids
- asthma
- COPD mechanisms
- COPD exacerbations
- COPD pathology
- COPD pharmacology
- cough/mechanisms/pharmacology
- respiratory infection
- viral infection
- asthma mechanisms
- asthma pharmacology
- emphysema
- primary pulmonary hypertension
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Cough
- sensory nerves
- transient receptor potential channels
- lung
- prostanoids
- asthma
- COPD mechanisms
- COPD exacerbations
- COPD pathology
- COPD pharmacology
- cough/mechanisms/pharmacology
- respiratory infection
- viral infection
- asthma mechanisms
- asthma pharmacology
- emphysema
- primary pulmonary hypertension
Key messages
What is the key question?
-
Endogenous mediators which are often elevated in respiratory disease states, such as PGE2 and bradykinin, are also known to cause cough. However, how this occurs is not known.
What is the bottom line?
-
Here we have elucidated the signaling mechanisms involved in this tussive response and identified a role for TRPA1 and TRPV1 channels.
Why read on?
-
These findings could have major implications for the treatment of cough which currently presents a significant unmet medical need.
Introduction
Cough is the most frequent reason for consultation with a family doctor,1 or with a general or respiratory physician. Patients with chronic cough probably account for 10–38% of respiratory outpatient practice in the USA.2 Chronic cough of various aetiologies is a common presentation to specialist respiratory clinics, and is reported as a troublesome symptom by 7% of the population.3 Treatment options are limited. A recent meta-analysis concluded that over-the-counter (OTC) cough remedies are ineffective4 and there is increasing concern about the use of OTC therapies in children. Despite its importance, our understanding of the mechanisms which provoke cough is poor.
The respiratory tract is innervated by sensory afferent nerves which are activated by mechanical and chemical stimuli.5 Activation of capsaicin-sensitive C-fibres and acid-sensitive, capsaicin-insensitive mechanoreceptors innervating the larynx, trachea, and large bronchi regulate the cough reflex.5 ,6 Endogenous inflammatory mediators are often elevated in respiratory disease states. For example, higher concentrations of prostaglandin E2 (PGE2)7 and bradykinin (BK)8 have been found in the airways of patients with asthma and chronic obstructive pulmonary disease. PGE2 and BK are also known to cause cough by stimulating airway sensory nerves.9 ,10 Furthermore, increased PGE2 levels have been found in idiopathic cough and cough associated with post-nasal drip, gastro-oesophageal reflux disease, cough variant asthma and eosinophilic bronchitis.11 It has previously been demonstrated that PGE2 activates guinea pig, mouse and human airway sensory nerves and causes cough via EP3 receptor activation.10 BK activates guinea pig airway sensory nerves and elicits cough via activation of the B2 receptor, but it is not known if the same process occurs in other species.9 Although we do have some information regarding which G-protein-coupled receptors (GPCRs) are activated by these endogenous tussive agents, it is still unclear what post-receptor signalling pathways are involved.
Recently, ion channels of the transient receptor potential (TRP) class such as TRPV1 have been implicated in the afferent sensory loop of the cough reflex12 ,13 and in the heightened cough sensitivity seen in disease.14 TRPA1 is a Ca2+-permeant non-selective channel with 14 ankyrin repeats in its amino terminus which also belongs to the larger TRP family. TRPA1 channels are activated by a range of natural products such as allyl isothiocyanate, allicin and cannabinol, found in mustard oil, garlic and cannabis15–17 and by environmental irritants (eg, acrolein, present in air pollution, vehicle exhaust and cigarette smoke),18–20 and is primarily expressed in small diameter, nociceptive neurons where its activation contributes to the perception of noxious stimuli such as itch.18 ,20 ,21 It has been demonstrated that stimulating TRPA1 channels activates vagal broncho-pulmonary C-fibres in rodent lung,22–24 inducing a late asthmatic response in sensitised rodents following allergen challenge25 and causing cough in guinea pig models and in normal human volunteers.26 Although many exogenous stimuli are known to activate TRPA1 and TRPV1, it is still unknown how cough and other reflexes are elicited in health and disease by endogenous agents, and whether these ion channels are involved. We hypothesised that the TRPA1 and TRPV1 ion channels may have a role as common effectors for such tussive agents.
Methods
Isolated vagal ganglia
Intracellular free calcium ([Ca2+]i) measurements were performed in dissociated jugular and nodose neurons. These studies were performed on all isolated vagal neurons (not airway specific), with the concentration–response data representing an overview of responding and non-responding cells. For subsequent antagonist studies only responding cells were analysed, with the criteria for a ‘responsive cell’ judged as an increase in [Ca2+]i of ≥10% of the K50 response. In each case, N = number of animals and n = number of cells tested. Comprehensive methods are detailed in online supplementary text.
Isolated vagus nerve preparation
Guinea pigs or mice (C57BL/6, Trpa1−/− and Trpv1−/− ) were sacrificed by injection of sodium pentobarbitone (200 mg/kg intraperitoneal injection). The vagus nerves were removed and experiments conducted in our fully characterised isolated vagus preparation, as described in previous publications.10 ,26 Human vagal tissue (n=6, two men, 27–72-year-old donors with no respiratory disease) was obtained from two sources—transplant tissue surplus to requirements (Harefield Hospital, UK); and purchased from the International Institute for the Advancement of Medicine (Edison, New Jersey, USA). In all cases, the tissue was consented for use in scientific research and ethics approval obtained from the Royal Brompton & Harfield Trust. See online supplementary text for full methods.
Conscious guinea pig cough model
Conscious unrestrained guinea pigs were placed in individual plastic transparent whole-body plethysmograph chambers (Buxco, Wilmington, North Carolina, USA) and cough detected as previously described.10 ,26
Data analysis and statistics
For imaging, RM is the maximum response observed expressed as a percentage of the K50 response. EC50 values quoted in the imaging studies are the concentrations of drug that produced 50% of the maximum response obtained. Inhibition of agonist responses in the isolated vagus nerve preparation was analysed by two-tailed paired t test, comparing responses to the agonist in the absence and presence of an antagonist in the same piece of nerve.
Inhibition of cough by TRPA1 and TRPV1 antagonists in vivo was analysed by Kruskal–Wallis test for multiple comparisons with Dunn's post hoc test, comparing responses from each group of antagonist/vehicle combination to the vehicle-only control. Data are presented as median ± IQR, with statistical significance set at p<0.05.
Results
Characterising agonist responses in isolated vagal ganglia
Capsaicin and acrolein produced concentration-related increases in [Ca2+]i in sensory neurons (supplementary figure 1A–D). PGE2 stimulation was multiphasic in both ganglia, of which 56.3% of jugular and 40% of nodose neurons responded. Overall, PGE2 increased [Ca2+]i in jugular neurons with an RM of 41±9% at 10 μM and an EC50 of 5.07±1.0 μM (N=5, n=16); whereas, in nodose neurons RM was only 11±2% at 10 μM with an EC50 of 3.11±0.4 μM (N=4, n=15). 52.9% of jugular neurons and 37.5% of nodose neurons responded to BK stimulation. Overall, BK induced 22±4% RM at 10 μM, with an EC50 of 2.32±0.36 μM in jugular neurons (N=5, n=17); and 17±3% RM at 30 μM with an EC50 of 2.2±0.2 μM in nodose neurons (N=5, n=24) (figure 1A–D).

Establishing concentration responses for prostaglandin (PGE2) and bradykinin (BK) in the in vitro preparations and in vivo cough model. (A–D) Concentration responses showing increases in intracellular calcium ([Ca2+]i) for PGE2 and BK in primary neurons isolated from guinea pig jugular (A, B) and nodose (C, D) ganglia. In each panel, histograms show an increase in [Ca2+]i for increasing concentrations of tussive agent. To take into account multiphasic shapes of some responses and their lengths, the calcium flux (area under curve (AUC)) generated by applications of tussive agents is normalised, and expressed as percentage of response to K50. The tussive agent used is indicated above each set of histograms and the concentration below each bar in μM (N=4–5, n=15–24). The trace in the lower left shows a typical recording of the light intensity over time following exposure to the agonist. Time and duration of drug application are indicated by a black bar above the trace. Time scale is given by the 1 min length-equivalent black bar shown below the trace. On the bottom right are display images taken during the recording. Time of the snapshot is indicated below each picture with zero being the start of tussive agent application. The pseudo colour code used for light intensity in the pictures is represented on the right of each set of images. (E, F) Perfusion for 2 min of BK (black bars) or PGE2 (white bars) activated (E) guinea pig and (F) mouse isolated vagus nerves in a concentration-dependent manner, measured as depolarisation of the nerve in mV (n=6). (G) The G-protein coupled receptor mediating BK-induced depolarisation (3 μM in guinea pig and 1 μM in mouse tissue) was identified as the B2 receptor in human (n=1–2) and guinea pig (n=6), and a combination of B1 and B2 receptors in the mouse (n=6) by incubating the nerve with either B1 (R715, 1 μM; checked bars) or B2 (WIN 64338, 10 μM; striped bars) selective antagonists for 10 min, measured as % inhibition of agonist responses. (E) BK (filled circles) and PGE2 (open circles) also induced concentration-related coughing in the conscious guinea pig, measured as the total number of coughs counted during 10 min of aerosol stimulation (n=4–8). Data are expressed as mean ± SEM of n observations (A–G) or median ± IQR (H). Statistical significance is indicated by *p<0.05 and **p<0.01, calculated as a paired t-test comparing responses in the same piece of nerve (human data were not analysed due to low numbers). Veh, vehicle. This figure is produced in colour in the online journal—please visit the website to view the colour figure.
Characterising agonist responses in vitro and in vivo
Capsaicin and acrolein produced concentration-related increases in depolarisation of guinea pig, mouse and human vagus nerve (online supplementary figure 1E–G). BK and PGE2 concentration dependently activated both guinea pig and mouse isolated vagus nerves, whereas the corresponding vehicles did not induce depolarisation (figure 1E,F). BK (3 μM) and PGE2 (10 μM) also activated human afferent sensory nerves (n=5–6, data not shown). The GPCR mediating the tussive effects of PGE2 has already been established as the EP3 receptor.10 Here, we show that BK activates only the B2 receptor in human and guinea pig, but B1 and B2 receptors in the mouse isolated vagus (figure 1G). It is possible that BK is inducing airway sensory afferent activation and cough via production of prostanoids.27 ,28 However, incubation of the vagus nerve with indomethacin did not alter BK-induced activation of either the guinea pig (20±11% inhibition, p>0.05) or wild-type mouse sensory nerves (13±10% inhibition; n=6, p>0.05; data not shown). The magnitude of BK-induced sensory nerve depolarisation was also similar in wild-type compared with EP3 −/− mouse vagus (n=6, p>0.05; data not shown), which is the GPCR through which PGE2 causes cough. Depolarisations to BK, PGE2, acrolein and capsaicin were abolished with the sodium channel blocker tetrodotoxin (n=3; 100% inhibition, data not shown). Further evidence for the observed depolarisation being mediated via sensory nerve activation comes from the in vivo experiments, demonstrating that BK and PGE2 successfully induce concentration-related coughing in conscious guinea pigs (figure 1H).
Characterising antagonist responses in isolated vagal ganglia
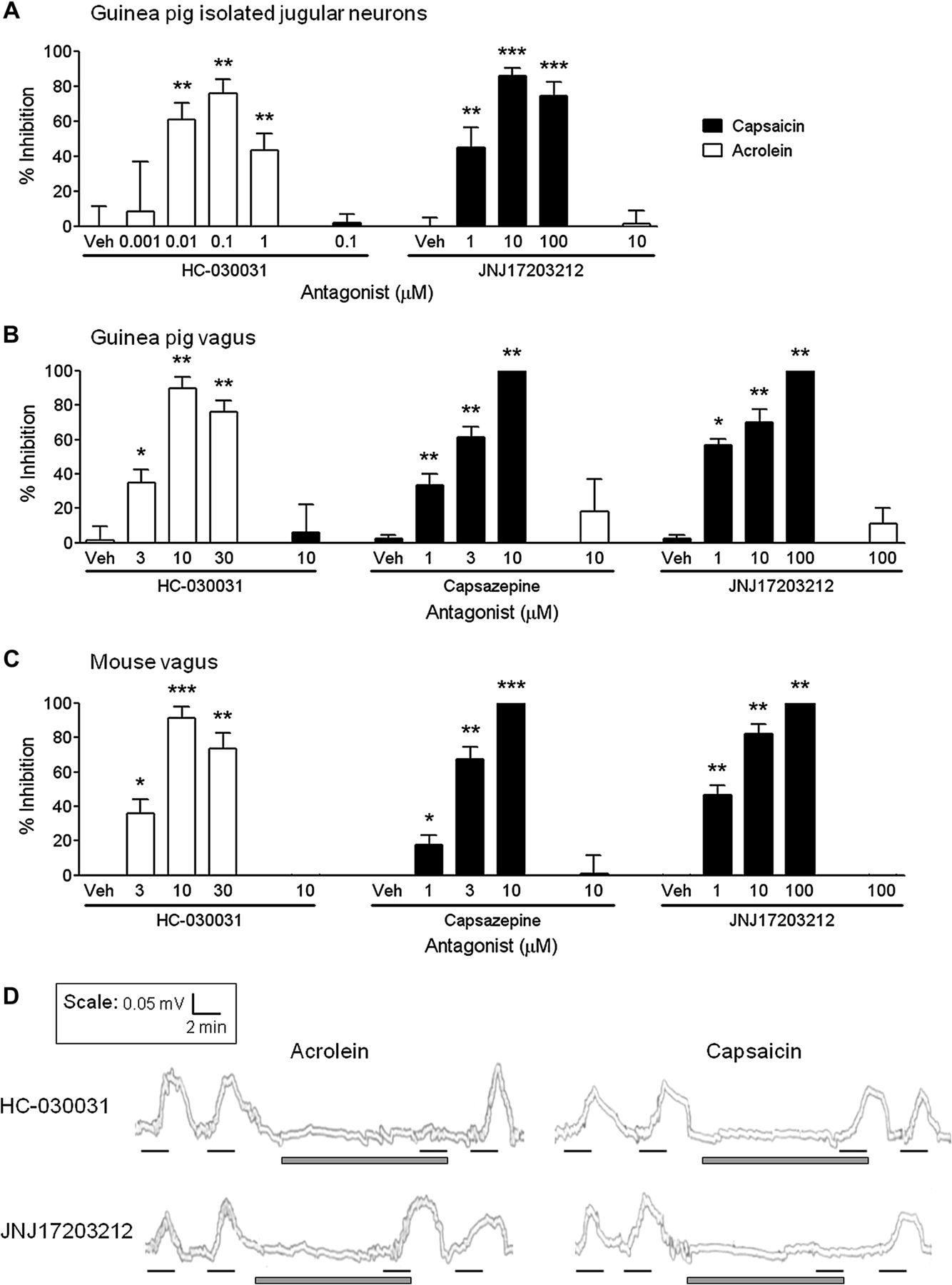
Concentration responses for the TRPV1-selective antagonist JNJ17203212 (JNJ) and TRPA1-selective antagonist HC-030031 (HC) were established in primary jugular cells for their ability to inhibit agonist-induced increases in [Ca2+]i (figure 2A). JNJ concentration-dependently inhibited increases in [Ca2+]i caused by the TRPV1-selective agonist capsaicin, with a maximal effect of 86±2% at 10 μM. Alternatively, HC concentration-dependently inhibited increases in [Ca2+]i induced by the TRPA1-selective agonist acrolein, with a maximal effect of 76±8% at 0.1 μM. At the concentration which caused maximal inhibition of its own receptor, 10 μM JNJ did not inhibit acrolein, and 0.1 μM HC did not inhibit capsaicin stimulation of jugular cells (figure 2A).
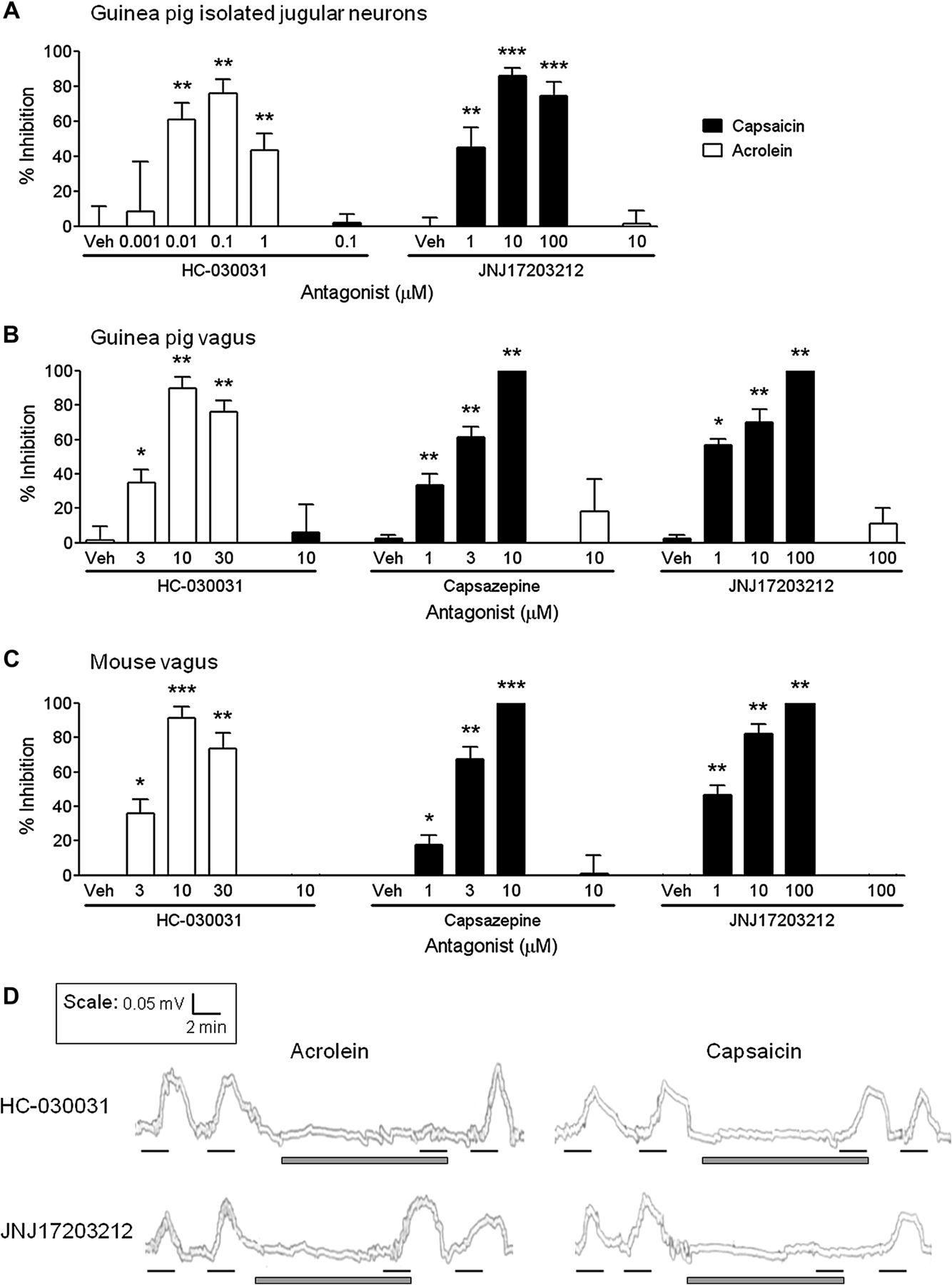
Characterisation of transient receptor potential channel A1 (TRPA1)-selective and TRPV1-selective antagonists in the in vitro primary ganglia and isolated vagus nerve preparations. The TRPA1 antagonist HC-030031 (HC) or TRPV1 antagonists JNJ17203212 (JNJ) or capsazepine (CAPZ) were assessed for their ability to inhibit capsaicin (black bars) and acrolein (white bars) responses in isolated guinea pig jugular neurons and guinea pig, mouse or human isolated vagus nerves. (A) HC concentration-dependently inhibited acrolein-induced (10 μM) increases in [Ca2+]i in guinea pig isolated jugular neurons, but had no effect on capsaicin (1 μM) at the concentration which maximally inhibited its own receptor (0.1 μM). Similarly, JNJ concentration-dependently inhibited capsaicin-induced responses, but had no effect on acrolein at 10 μM (N=3–4, n=5–19). (B, C) HC concentration-dependently inhibited acrolein-induced (300 μM) depolarisation of the guinea pig and wild-type mouse isolated vagus nerves, but had no effect on capsaicin (1 μM) stimulation. Conversely, TRPV1 antagonism with capsazepine or JNJ17203212 concentration-dependently inhibited capsaicin-induced depolarisation in guinea-pig and mouse isolated vagus nerves, but had no effect on acrolein stimulation at 10 μM or 100 μM, respectively (n=6). (D) Representative traces showing inhibition of human vagus nerve depolarisation with 10 μM HC when stimulated with acrolein (300 μM) but not capsaicin (1 μM). Conversely, 100 μM JNJ inhibited capsaicin but not acrolein responses (n=2–3). Black lines represent agonist incubation (2 min) and grey bars antagonist incubation (10 min). Data are presented as mean ± SEM of n observations, calculated as % inhibition of agonist responses. *(p<0.05), **(p<0.01) and ***(p<0.0001) indicate statistical significance, paired t-test comparing responses in the same piece of nerve. Veh, vehicle for the antagonist (0.1% dimethyl sulfoxide).
Characterising TRP-selective antagonists in vitro
Depolarisation of guinea pig and mouse vagus nerve by acrolein was concentration-dependently inhibited with the TRPA1-selective antagonist HC. Similarly, capsaicin responses were concentration-dependently inhibited by the TRPV1-selective antagonists capsazepine (CAPZ) and JNJ (figure 2B,C). At the concentration which maximally inhibited acrolein, HC (10 μM) did not inhibit capsaicin-induced nerve depolarisation; and equally CAPZ (10 μM) and JNJ (100 μM) did not inhibit acrolein-induced nerve depolarisation (figure 2B,C). This suggests that these compounds are not exhibiting off-target actions at these concentrations. Subsequently, the effects of HC (10 μM) and JNJ (100 μM) were investigated in human isolated vagus. In these experiments (n=2–3), acrolein responses were abolished by HC but not affected by JNJ; whereas, capsaicin responses were abolished by JNJ but not affected by HC (example traces shown in figure 2D). Vehicle control (0.1% dimethyl sulfoxide (DMSO) vol/vol) did not inhibit agonist responses (data not shown).
Determining the role of TRPA1 and TRPV1 in PGE2 and BK induced vagal ganglia and sensory nerve activation in vitro
Having characterised the available tools, and confirmed selectivity of the antagonists, the role of TRPA1 and TRPV1 in PGE2-induced and BK-induced vagal ganglia and nerve stimulation was established. HC (0.1 μM) or JNJ (10 μM) partially inhibited PGE2-induced (55±4% and 40±9%, respectively) or BK-induced (45±5% and 46±7%, respectively) increases in [Ca2+]i in guinea pig primary cells isolated from jugular vagal ganglia (p<0.01). Furthermore, when used in combination, HC and JNJ inhibited PGE2-induced [Ca2+]i elevation by 88±3% and BK by 80±12% (p<0.0001). In contrast, vehicle incubation had no effect on [Ca2+]i (−1±11% for PGE2 and −4±8% for BK; p>0.05) (figure 3A,B).
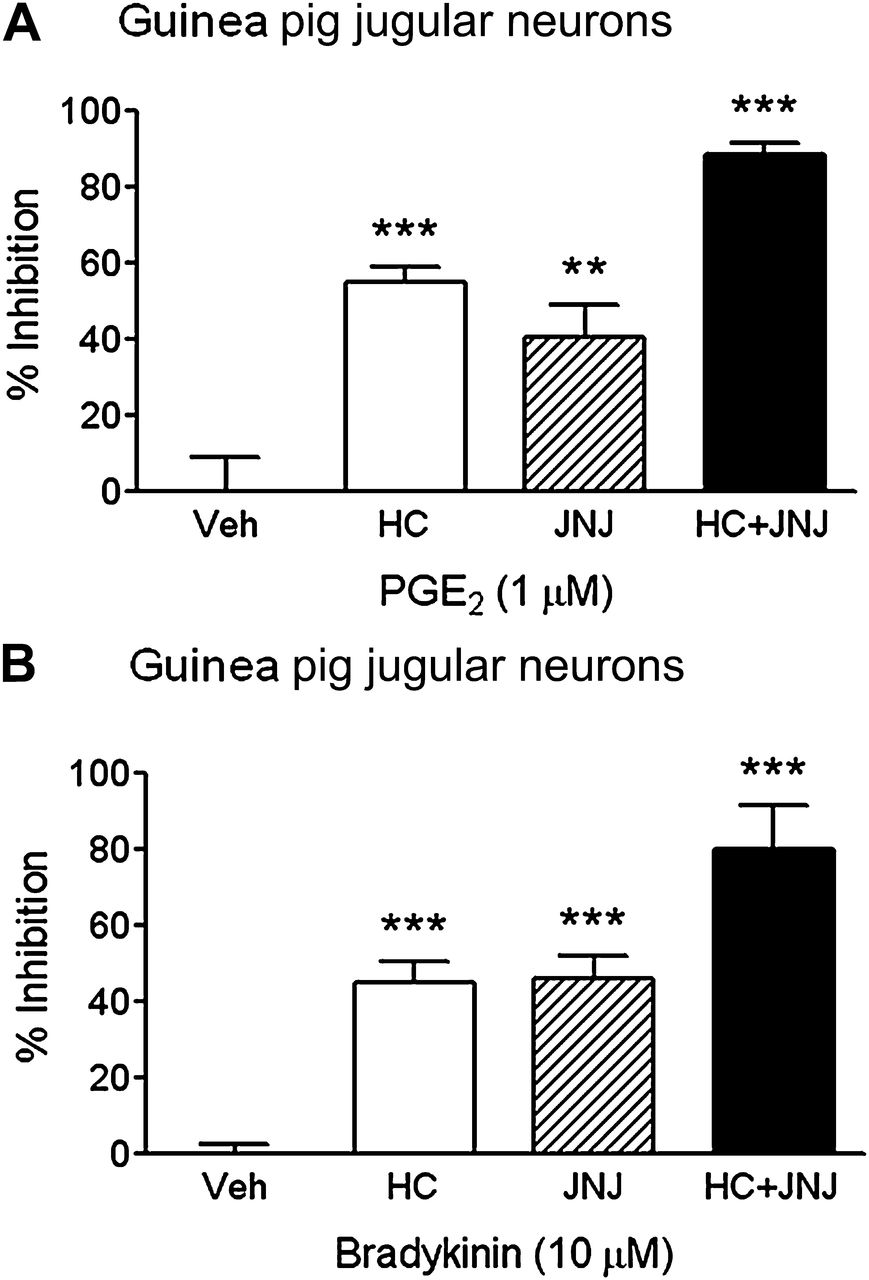
Determining the role of transient receptor potential channel A1 (TRPA1) and TRPV1 in prostaglandin E2 (PGE2) and bradykinin (BK) induced isolated primary jugular neurons. The TRPA1 antagonist HC-030031 (HC, 0.1 μM; white bars); TRPV1 antagonist JNJ17203212 (JNJ, 10 μM; striped bars); and a combination of HC+JNJ (black bars) were assessed for their ability to inhibit (A) 1 μM PGE2 and (B) 10 μM BK responses in isolated guinea pig jugular neurons. HC or JNJ partially inhibited PGE2 and BK responses, whereas HC+JNJ almost completely abolished increases in [Ca2+]i. Data are presented as mean ± SEM of N=3–5, n=10–19 observations, calculated as % inhibition of agonist responses. **(p<0.01) and ***(p<0.0001) indicate statistical significance, paired t-test comparing responses in the same neuron. Veh, vehicle for the antagonist (0.1% dimethyl sulfoxide).
TRPA1 antagonism with HC partially inhibited PGE2 and BK responses in the guinea pig isolated vagus nerve (44±5% and 47±3%, respectively). Additionally, TRPV1 antagonism with CAPZ or JNJ also partially inhibited PGE2 (45±3% and 48±4%, respectively) and BK responses (55±3% and 49±8%, respectively) (p<0.05). When used in combination, HC+JNJ abolished vagus nerve responses to PGE2 (90±4%) and BK (95±4%) (p<0.0001; figure 4A,B). To further confirm these results the experiments were repeated in a second species. The mouse was chosen because we have access to Trpa1−/− and Trpv1−/− mice, and have previously shown that mouse vagus responds in a similar fashion to human isolated vagus.10 ,26 Knockdown of the TRPA1 or TRPV1 gene was confirmed by genotyping (figure 4C). Vagal nerve activation induced by acrolein and capsaicin were initially assessed to ensure phenotypical loss of TRPA1 and TRPV1 responses (data not shown). The results obtained in guinea pigs were then confirmed by comparing the magnitude of stimulation of the endogenous tussive agents in wild-type mice to that of Trpa1−/− and Trpv1−/− animals. The Trpa1−/− and Trpv1−/− responses to PGE2 and BK stimulation were approximately half those seen in wild-type mouse vagal tissue (p<0.01; data not shown).
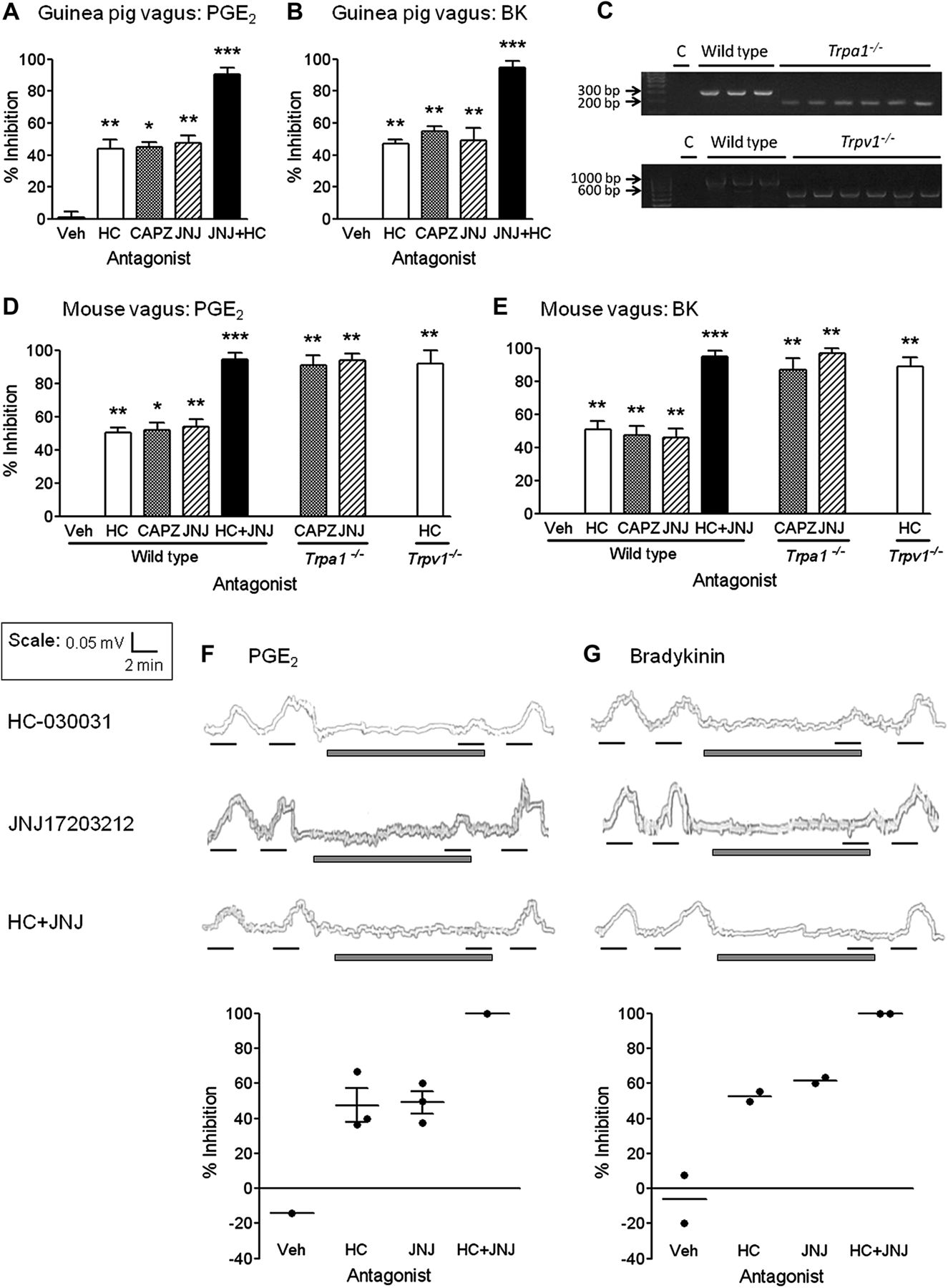
Determining the role of transient receptor potential channel A1 (TRPA1) and TRPV1 in prostaglandin E2 (PGE2) and bradykinin (BK) induced sensory nerve activation. The TRPA1 antagonist HC-030031 (HC 10 μM; white bars), TRPV1 antagonists capsazepine (CAPZ 10 μM; grey bars) and JNJ17203212 (JNJ 100 μM; striped bars), and a combination of HC+JNJ (black bars) were assessed for their ability to inhibit PGE2 (10 μM) and BK (3 μM in guinea pig and human, and 1 μM in mouse tissue) isolated vagus nerve responses. (A, B) HC, CAPZ or JNJ partially inhibited PGE2 and BK responses in isolated guinea pig vagus tissue, whereas, HC+JNJ almost completely abolished nerve activation. (C) Knockdown of the TRPA1 or TRPV1 gene was verified by genotyping. Bands were expected at 317 bp for wild-type and 184 bp for Trpa1−/− ; and 984 bp for wild-type and 600 bp for Trpv1−/− mice. C, water (negative control); bp, base pair. (D, E) HC, CAPZ or JNJ partially inhibited PGE2 and BK responses in isolated wild-type mouse vagus tissue, whereas, HC+JNJ almost completely abolished nerve activation. In agreement with this, sensory nerves taken from genetically modified mice Trpa1−/− or Trpv1−/− tested in combination with the alternative TRPV1 or TRPA1 antagonist also largely eliminated sensory nerve responses to PGE2 and BK. (F, G) HC and JNJ partially inhibited PGE2 and BK responses in human isolated vagal tissue, whereas, HC+JNJ abolished nerve depolarisation. Example traces are shown above, where black lines represent agonist incubation (2 min), and grey bars represent antagonist incubation (10 min). Scatter plots of % inhibition are shown below and time and magnitude scales for the traces are shown in the top left hand corner. Data are presented as mean ± SEM of n=6 observations for guinea pig and mouse experiments, and n=2–3 for human experiments, calculated as % inhibition of agonist responses. *(p<0.05), **(p<0.01) and ***(p<0.0001) indicate statistical significance, paired t-test comparing responses in the same piece of nerve. Veh, vehicle for the antagonist (0.1% dimethyl sulfoxide).
In agreement with the results obtained from guinea pig vagus, wild-type mouse nerve responses to PGE2 and BK were partially inhibited by HC (47±2% and 51±5%, respectively), CAPZ (49±4% and 48±6%, respectively) and JNJ (54±5% and 46±5%, respectively) (p<0.05). When used in combination, HC+JNJ abolished vagus nerve responses to PGE2 (94±4%) and BK (95±3%) (p<0.0001; figure 4D,E). And when tissue taken from genetically modified animals was tested in combination with the alternative antagonist, responses were again virtually abolished. In Trpa1−/− tissue PGE2 was inhibited 91±6% and 94±4%; and BK 87±7% and 97±3% by CAPZ and JNJ, respectively (p<0.01; figure 4D,E). In Trpv1−/− tissue, PGE2 was inhibited 92±8% and BK 89±6% by HC (p<0.01; figure 4D,E).
We were able to confirm the above results in human vagal tissue (n=2–3). In these experiments, both PGE2 and BK were partially inhibited by either HC or JNJ antagonism with complete inhibition when the antagonists were used in combination. Vehicle control (0.1% DMSO vol/vol) did not inhibit agonist responses (figure 4F,G).
Determining a role for TRPA1 and TRPV1 in PGE2 and BK induced guinea pig cough
Concentration responses for the selective agonists capsaicin (TRPV1; figure 5A) and acrolein (TRPA1; figure 5B) were initially established to determine a submaximal concentration for which to test the antagonists (60 μM capsaicin and 100 mM acrolein). Subsequently, the maximally effective doses of the selective antagonists HC (TRPA1) and JNJ (TRPV1) which did not display off-target effects on the alternative receptor were determined. HC dose-dependently inhibited acrolein-induced cough in vivo, but at 300 mg/kg had no effect on capsaicin; and JNJ concentration-dependently inhibited capsaicin-induced cough but at 100 mg/kg had no effect on acrolein in conscious guinea pigs (figure 5C,D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Determining the role of transient receptor potential channel A1 (TRPA1) and TRPV1 in prostaglandin E2 (PGE2) and bradykinin (BK)-induced cough in conscious guinea pigs. (A) Capsaicin and (B) acrolein concentration-dependently induced coughing in conscious, unrestrained guinea pigs. Tussive agents were aerosolised for 5 min, the number of coughs was counted during this time and for a further 5 min post stimulation (10 min total). Data are presented as mean ± SEM of n=10–12 observations. (C, D) Animals received intraperitoneal injections with a concentration of TRPA1 antagonist HC-030031 (HC), TRPV1 antagonist JNJ17203212 (JNJ) or vehicle (Veh) 1 h prior to 5 min aerosol stimulation with a tussive agonist. The number of coughs was counted during the 5 min stimulation plus a further 5 min (10 min total). (C) HC concentration-dependently inhibited acrolein-induced coughing (100 mM; open circles), but had no effect on capsaicin cough (60 μM; filled circles) at 300 mg/kg. (D) Conversely, JNJ concentration-dependently inhibited capsaicin-induced cough, with no effect on acrolein at 100 mg/kg. Data are presented as mean ± SEM of n=8–10 observations. (E, F) Animals received intraperitoneal injection with HC (300 mg/kg; filled circles), JNJ (100 mg/kg; filled squares), a combination of both antagonists (HC+JNJ; filled triangles), or appropriate Veh (open circles) 1 h prior to stimulation with a tussive agonist. (E) PGE2 (300 μg/ml) or (F) BK (3 mg/ml) were aerosolised for 10 min, during which time the number of coughs was counted. Compared with vehicle control, pretreatment with either HC or JNJ significantly inhibited PGE2-induced or BK-induced coughing; and pr-treatment with HC+JNJ abolished cough altogether. Data are presented as median ± IQR of n=10–12 observations. *(p<0.05), **(p<0.01) and ***(p<0.0001) indicate statistical significance, Kruskal–Wallis one-way analysis of variance with Dunn's multiple comparison post-test.
The selective antagonists were then tested against PGE2-induced and BK-induced cough, appropriate concentrations of which had been determined earlier (300 μg/ml PGE2 and 3 mg/ml BK) (figure 1H). The in vivo guinea pig cough results agree with the in vitro findings. When pretreated with vehicle control, PGE2 induced coughs in response to 10 min aerosol stimulation. This was reduced with either HC or JNJ pretreatment, respectively. Similarly, pretreatment with HC or JNJ antagonists reduced BK-induced coughing compared with vehicle control (figure 5E,F). When pretreated with a combination of HC+JNJ, the cough responses to PGE2 and BK were completely abolished (figure 5E,F).
Discussion
Despite its importance, our understanding of the mechanisms which provoke cough and the endogenous tussive agents involved in health and disease is poor. Chronic cough is often associated with an underlying inflammatory condition, as in asthma and chronic obstructive pulmonary disease, but the endogenous mediators and signal transduction pathways which initiate cough are not known. Inflammatory diseases are associated with enhanced release of inflammatory mediators in the airways.7 ,8 Two such mediators are PGE2 and BK, which have been shown to induce coughing in humans29 ,30 and animals.9 ,10 Interestingly, cough associated with patients who take angiotensin-converting enzyme (ACE) inhibitors has also been suggested to be due to the increased levels of bradykinin.9 Furthermore, PGE2 levels have been found to be elevated in induced sputum of patients with chronic cough.11 It has previously been demonstrated that PGE2 activates guinea pig, mouse and human airway sensory nerves and evokes cough in guinea pigs via the EP3 receptor.10 However, BK stimulates guinea pig sensory nerves and elicits cough via activation of the B2 receptor.9 ,31 In these studies we demonstrate that BK and PGE2 are able to activate sensory jugular ganglia; depolarise guinea pig, mouse and human vagal afferents; and evoke cough in a guinea pig model in a concentration-related fashion. Interestingly, the bradykinin B2 receptor mediated sensory nerve activation in the isolated guinea pig and human vagal nerve assays but the B1 receptor also played a role in the BK-induced activation of the mouse vagus, highlighting a species difference.
To induce coughing, post-receptor signalling pathways downstream of GPCR coupling are likely to cause the opening of membrane-bound ion channels leading to activation of airway sensory nerves and subsequent coughing. Previously other groups have presented data implicating the TRP family of ion channels in sensory nerve activation and the cough reflex elicited by BK.32 In these studies we have confirmed these data and extended these findings by confirming a partial inhibition of BK-induced sensory nerve activation by TRPV1 antagonists. Similar results were obtained with PGE2, with the TRPV1 antagonists producing partial inhibition of vagal sensory nerve activation in all species, guinea pig jugular neurons, and in a guinea pig cough model. At concentrations which were selective for inhibition of TRPA1 ligands (and not TRPV1 ligands) we also found that the TRPA1 antagonist, HC-030031 inhibited BK-induced and PGE2-induced [Ca2+]i in jugular neurons, vagal sensory nerve activation and cough. Furthermore, both antagonists together (JNJ17203212 and HC-030031) completely inhibited PGE2 and BK in vitro and in vivo responses. In vitro pharmacological sensory nerve studies were confirmed in tissue from Trpv1−/− and Trpa1−/− gene deleted mice.
The mechanisms downstream of GPCR coupling that lead to either sensitisation or activation of ion channels are not yet fully understood, but phospholipase C (PLC) and protein kinase A (PKA) pathways are thought to be important in the signalling for a number of TRP channels.32–34 GPCR binding to Gq-coupled receptors can lead to activation of PLC, hydrolysis of phosphatidylinositol-(4,5)-biphosphate (PIP2) to yield inositol-(1,4,5)-triphosphate (IP3), production of diacylglycerol (DAG) and activation of phosphokinase C (PKC). PKC and DAG have been found to directly bind the TRPV1 receptor; and IP3-induced release of intracellular calcium stores may be involved in activation of TRPA1. Moreover, PIP2 is thought to constitutively inhibit TRP receptors. Therefore, its hydrolysis by PLC may disinhibit these ion channels, sensitising them to subsequent stimulation.34 Alternatively, PKA-dependent phosphorylation can occur through activation of Gs-coupled receptors, thereby enhancing ion channel excitability.34
A number of the functional responses elicited by BK are caused via indirect effects, including the release of other endogenous mediators downstream of arachadonic acid. We established here that the stimulatory effects of BK on isolated vagus nerves were not due to subsequent release of prostanoids by using the general cyclo-oxygenase inhibitor indomethacin. However, a number of studies have also implicated downstream release of lipoxygenase products. For example, BK evokes the release of 15-HETE from airway epithelial cells.27 Furthermore, 12-lipoxygenase and 5-lipoxygenase products have been implicated in BK-induced stimulation of airway afferent nerve terminals via TRPV1 channel activation.35 It is therefore plausible that BK may be causing cough via the release of lipoxygenase products downstream of arachadonic acid.
In this paper, we have used in vitro cellular and tissue preparations, and an in vivo animal model to investigate the cough reflex. The data generated with these models are useful in attempting to understand cough; however, each model has its limitations. In the calcium-imaging preparation, we cannot determine if there are phenotypical changes induced in the primary ganglia cells during the isolation process. One of the benefits of the isolated vagus nerve preparation is that we can parallel our animal experiments in human tissue. However, the agents being tested are applied to the axon of the vagus nerve (not the nerve endings), meaning that the extracellular depolarisation signal recorded represents a summation of the change in membrane potential of all the nerve fibres being carried by the vagus. In addition, receptor expression and signal transduction mechanisms may differ from those at the peripheral endings. Finally, though the conscious guinea pig cough model is generally considered to be a valid tool for studying the cough reflex, there are a number of reported differences between the actions of certain drugs in guinea pig and man. These differences could be due to strong tachykinin-driven responses via sensory nerves in the guinea pig airways or because potential anti-tussives have been trialled in clinical studies in which cough was not the primary endpoint and where there was no objective cough monitoring and so any efficacy may have been hard to capture. Furthermore, in guinea pig studies compounds are often not dose limited as they are in the clinic due to safety concerns.
The findings presented here are important for our understanding of the cough reflex (and in particular inflammatory and ACE inhibitor induced cough) and strongly support a role for TRPA1 and TRPV1 as common effectors of the tussive response to endogenous tussive agents. These studies were conducted in tissues and in vivo models under ‘normal’ physiological conditions and so a role for these TRP channels has not been established under pathophysiological conditions. However, it has been shown that patients suffering from chronic cough exhibit an increased TRPV1 expression within the lungs, which was correlated with an increase in cough sensitivity to capsaicin challenge,14 indicating that TRP channels could be common effectors of tussive responses in disease and that these channels could be associated with long-term potentiation of the cough reflex. Studies have not yet been conducted to show if TRPA1 is overexpressed in pathological cough in man because suitable antibodies are not available but these will be important studies to perform when appropriate tools are developed. Current research investigating the pathogenesis of cough supports the development of TRP channel inhibitors as novel and selective treatment modalities.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online materials 1
- Data supplement 2 - Online materials 2
- Data supplement 3 - Online figure 1
Footnotes
-
Additional materials are published online only. To view these files please visit the journal online (http://dx.doi.org/10.1136/thoraxjnl-2011-201443).
-
Funding MAB, SAM and MG were funded by project grants from the Medical Research Council (MRC, UK) (MAB, G0800196; SAM, MG, G0800195). The human tissue experiments in this study were undertaken with the support of the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. ED was funded by a Wellcome Trust project grant (089301/Z/09/Z).
-
Competing interests None.
-
Ethics approval Ethics approval was provided by Royal Brompton & Harefield Trust.
-
Provenance and peer review Not commissioned; externally peer reviewed.