Article Text

Abstract
Cystic fibrosis (CF), the most common life-shortening genetic disorder in Caucasians, affects approximately 70 000 individuals worldwide. In 1998, the Cystic Fibrosis Foundation (CFF) launched the CF Therapeutics Development Network (CF-TDN) as a central element of its Therapeutics Development Programme. Designed to accelerate the clinical evaluation of new therapies needed to fulfil the CFF mission to control and cure CF, the CF-TDN has conducted 75 clinical trials since its inception, and has contributed to studies as varied as initial safety and proof of concept trials to pivotal programmes required for regulatory approval. This review highlights recent and significant research efforts of the CF-TDN, including a summary of contributions to studies involving CF transmembrane conductance regulator (CFTR) modulators, airway surface liquid hydrators and mucus modifiers, anti-infectives, anti-inflammatories, and nutritional therapies. Efforts to advance CF biomarkers, necessary to accelerate the therapeutic goals of the network, are also summarised.
- Cystic Fibrosis
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Structure of the network, CFF partnership, industry partnerships, operational issues that foster success
Cystic fibrosis (CF), the most common life-shortening genetic disorder in Caucasians, affects approximately 70 000 individuals worldwide and meets the US Food and Drug Administration (FDA) definition of an orphan disease.1 Historically, the limited number of patients at each clinical site led to underpowered research studies2 and was a critical barrier to development and regulatory approval of new therapies. Hence, the Cystic Fibrosis Foundation (CFF) launched the CF Therapeutics Development Network (CF-TDN) in 1998 to accelerate the clinical evaluation of new CF therapies.3 ,4 From its inception, the CF-TDN was organised to provide necessary infrastructure to promote efficient study conduct. In addition, patient safety has always been the highest priority.3 From the beginning, the programme was composed of Therapeutic Development Centers (TDCs) across the USA, a clinical and data coordinating centre located at Seattle Children's Research Institute, and a number of resource centres with expertise in interpretation or performance of specialised outcomes measures (microbiology, imaging, infant lung function testing, inflammatory mediators, sweat testing, CF transmembrane conductance regulator (CFTR) detection and cytology interpretation) (see online supplementary figure S1). To maintain scientific integrity and safety of CF-TDN studies, there is a Steering Committee, protocol review of all multicenter trials, and review of presentations and publications from CF-TDN studies (see online supplementary figure S2). The CFF-supported Data Safety Monitoring Board located at the University of Arizona has oversight of clinical trials at all centres. The TDCs are selected by a competitive, peer-reviewed application process from the 120 US CF care centres4 ,5 and receive infrastructure support for a research coordinator and other necessary staff. As a result, TDCs have been able to retain well trained, experienced research staff teamed with site investigators. Over the past 15 years, the CF-TDN has successfully completed over 75 clinical trials along the entire developmental path from phase I to phase III registration trials. The goal to bring new therapies to the CF community has been achieved with FDA approval of several therapies (table 1).6 A key factor in the success of the CF-TDN has been its partnership with industry, providing a full range of consultative services from the earliest discussions about protocol development through critical protocol review to feedback on the industry/contract research organisation experience from the study site perspective. Above all, industry sponsors value the CF-TDN's experience in the conduct of CF clinical research in the USA.
CF therapeutics evaluated by the TDN and approved by the FDA
The coordinated efforts of the CF-TDN also include the development of novel outcome measures (below), use of banked specimens and data to conduct ancillary studies,7 ,8 and development of novel approaches to study design and analyses.9 In addition, the network has focused on a quality improvement (QI) initiative based upon groundbreaking work of the CF care centre programme.5 ,10 The need for improved communications and a QI programme became essential when the CF-TDN expanded from 8 to 77 TDC sites from 1998 to 2009, encompassing a population of 19 000 patients. Communication was improved using quarterly newsletters, conference calls, annual in-person meetings, and the creation of the CF-TDN website, CF Clinical Research Net, which tracks site study activity, provides tools for site training and programme enhancement, hosts committee work, posts news and information regarding studies and network programmes, and provides access to the network database. A commitment to QI for all aspects of clinical studies has paralleled the growth in network size. Here we summarise recent progress of the network across key therapeutic areas (see CF-TDN Pipeline depicted on the front cover).
CFTR modulators
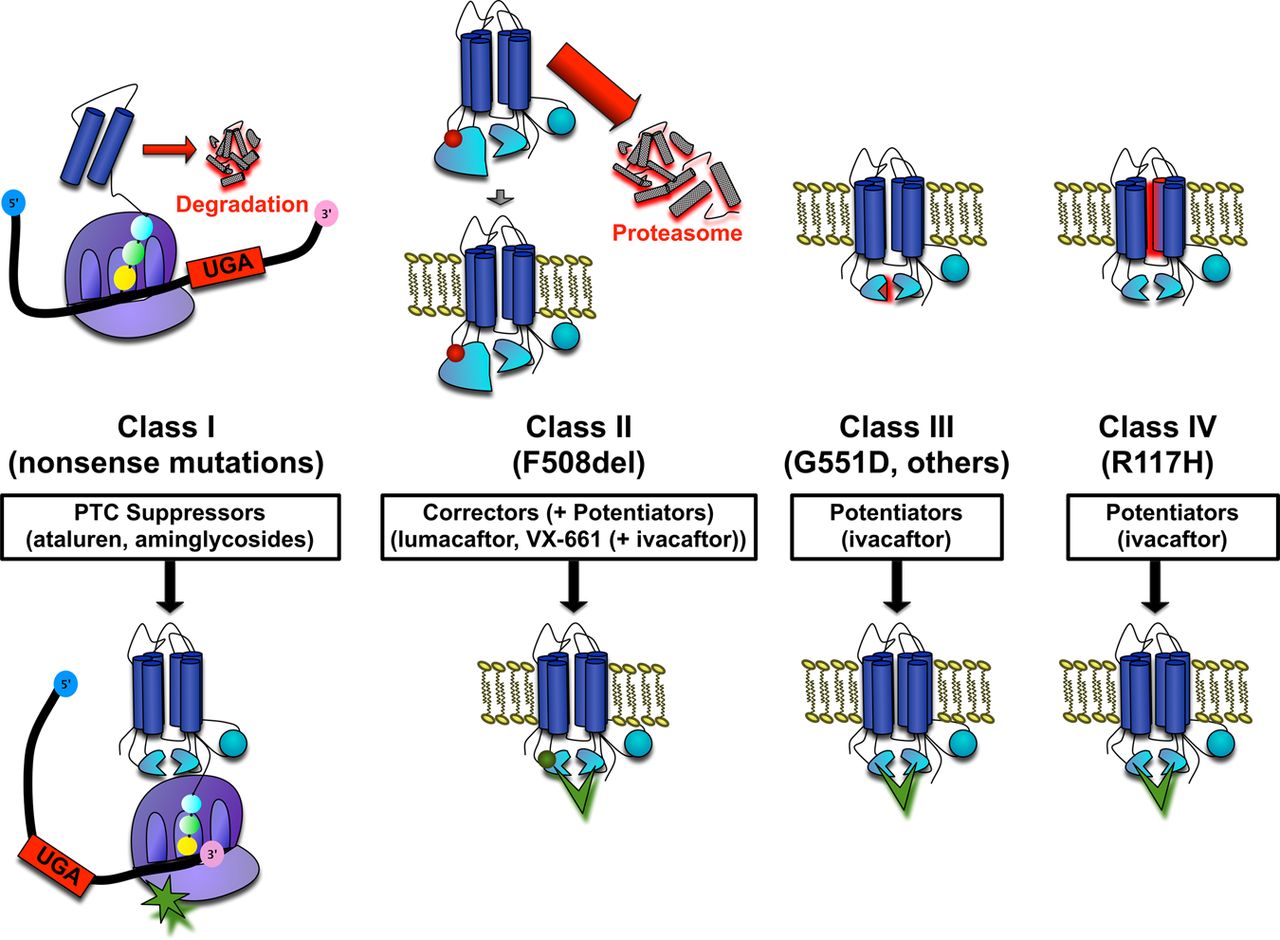
CFTR modulators have brought a personalised approach to altering the basic defect in the disease using gene class specific therapy (see reviews11–15). By grouping CFTR mutations into causative classes, specific approaches can be developed that address the underlying molecular defect on an individual patient level (figure 1), and serves as a paradigm for the treatment of other genetic diseases.16 The evaluation of these novel agents required expertise in proof-of-concept clinical trial design, biomarkers of pharmacological activity, and clinical outcome measures in an entirely new therapeutic area, and were promoted by close partnerships between the pharmaceutical industry and the CFF. Recent results have firmly established that rescue of CFTR-mediated anion transport can result in marked clinical benefit if CFTR functional improvement is sufficient,6 a concept likely to be a major contributor to CF-TDN efforts in the future.
{kind=link}
Cystic fibrosis transmembrane conductance regulator gene mutation classes and therapeutic approaches under study by the cystic fibrosis Therapeutics Development Network (CF-TDN). Note: approaches to class V (splicing mutations that alter surface CFTR levels), and class VI (mutations in the PDZ binding domain that decrease surface stability and/or alter recycling) CFTR mutations are under development, but have not yet entered clinical testing through the TDN.
CFTR modulator therapies have been directed towards specific disease-causing mutations and the molecular pathways that underlie their cause.12 ,16 A crucial phase II trial that tested the CFTR potentiator ivacaftor (Kalydeco, VX-770, Vertex Pharmaceuticals, Boston, MA, USA) in 40 patients with CF with at least one copy of the G551D mutation, a relatively common class 3 gating mutant, in a two-part randomised placebo-controlled design was the first to definitively establish that CFTR functional rescue detected by simultaneous improvement in nasal potential difference (NPD) and sweat chloride could result in meaningful changes in lung function.17 This was rapidly followed by two phase III trials in patients with G551D CF to establish the clinical effect of ivacaftor during a sustained period. In a trial in older children and adults (aged 12 and above), forced expiratory volume in 1 s (FEV1) improved 10.5% at 24 weeks, an effect durable at 48 weeks and accompanied by a 55% reduction in the probability of experiencing a pulmonary exacerbation, a 3.1 kg weight gain (compared with 0.9 kg in the placebo group), and improved respiratory symptoms as assessed by the CFQ-R, a patient reported quality-of-life index.6 As with phase II testing, sweat chloride improved by around 48 mEq/litre, and mean concentration was around 55 mEq/litre, a level below the traditional diagnostic threshold of CF (60 mEq/litre). Similar results were reported in a study of patients with G551D aged 6–12,18 enabling prompt approval of the drug for patients with G551D CF aged 6 and above by the FDA and European regulatory authorities. The CF-TDN will support additional studies to evaluate the safety and pharmacokinetics of ivacaftor in younger patients with CF (aged 2–5), which could also begin to assess the effects of early CFTR rescue on the pancreas. The rapid approval and availability of ivacaftor has also enabled the G551D observational study (GOAL), which will assess the clinical effects of ivacaftor in patients with G551D CF post-approval, including the response of several mechanistic biomarkers such as sweat rate, mucociliary clearance imaging, gastrointestinal pH, and sputum measures of inflammation and microbiology; these studies are intended to advance our understanding of the effects of efficacious CFTR modulation on disease-relevant biomarkers and pathways. Since ivacaftor also exhibits robust activity in other rare but clinically relevant CFTR gating mutations, the drug is also being studied in patients with CFTR gating mutations other than G551D. Ivacaftor will also be tested in the archetype conductance mutation R117H, which could establish whether potentiation of CFTR gating is sufficient to partially ameliorate non-gating mutations, setting the stage for other studies involving rare mutations localised to the cell surface.
Since ivacaftor alone is not sufficient to meaningfully alter CFTR activity in patients with CF homozygous for F508del,19 a class 2 mutant and the most common CFTR mutation, ‘correctors’ of CFTR misfolding are required to augment localisation of the F508del CFTR mutation to the cell surface. The CFTR correctors lumacaftor (VX-809, Vertex Pharmaceuticals, Cambridge, Massachusetts, USA) and VX-661 (Vertex Pharmaceuticals) have entered clinical testing within the CF-TDN for this purpose. While monotherapy with lumacaftor in adults homozygous for the F508del mutation only modestly reduced sweat chloride (8 mEq effect vs placebo at the highest dose tested (200 mg)) and did not improve lung function or NPD, evidence of sweat chloride improvement, albeit small in magnitude, suggested correction of the F508del CFTR protein was possible.20 To improve upon the effects of a corrector alone, lumacaftor is presently being tested at higher doses in a three-part study alone and in combination with ivacaftor, thereby evaluating the effects of augmenting CFTR channel gating following partial restoration of CFTR to the cell surface, a strategy justified by preclinical research21 ,22 and the known effects of the F508 deletion on gating in addition to cellular processing.23 Interim results through part 2 of the study have indicated that high doses of lumacaftor combined with ivacaftor improves FEV1 by 7.4%, and have recently provided the basis for a pivotal programme to evaluate combination therapy in patients with CF homozygous for F508del that will necessitate involvement in a many sites, including those beyond the network infrastructure. TDCs are also involved in testing the CFTR corrector VX-661 alone and in combination with ivacaftor as an alternate CFTR corrector to lumacaftor. In total, the combination of CFTR potentiators and correctors could result in effective therapies in a large proportion of patients with CF (table 2).
CFTR-based therapies completed or in progress within the Therapeutics Development Network
The CFTR modulator class also includes treatment strategies directed towards premature termination codons (class 1 mutations). Following the discovery that aminoglycoside antibiotics can induce readthrough premature termination codons resulting in full-length functional protein,24 an initial TDN network study focused on topical administration of aminoglycosides25; while bioactivity was not detectable and conflicted with other single-centre studies,26–28 this trial provided experience evaluating CFTR modulators helpful for subsequent studies, including those evaluating the small molecule ataluren (PTC124, premature termination codon therapeutics). Based on efficacy in vitro29 and in animal models,30 ataluren was studied in a series of conflicting phase II trials,31–33 one of which involved the TDN network.32 Results of a long-term study showed no improvement in FEV1 % predicted, the primary endpoint, but did demonstrate a small effect on lung function in a predefined subset of individuals who were not treated with inhaled antibiotics, which can alter the efficacy of ataluren.34 While the clinical status of ataluren remains uncertain at present, given the strong foundation of translational readthrough as a therapeutic strategy for CF and many other genetic diseases, studies involving this compound and other readthrough approaches within the CF-TDN are likely in the future.
Airway surface liquid depth and mucus modification
The rationale supporting the development of therapeutics targeting airway surface liquid (ASL) volume and mucus properties comes from basic research on the pathogenesis of CF lung disease.35 Numerous experimental models support the central role of CFTR as both a chloride channel and as a regulator of other ion channels in airway epithelia.36 ,37 Disruption of CFTR activity, in turn, has been linked to the development of defective mucus clearance via depletion of ASL volume and mucus dehydration.38 These and perhaps other distinct effects on airways defence may ultimately explain the initial, profound vulnerability to chronic bacterial airways infection that exists in CF, while also contributing to disease progression.
Approaches to increase fluid secretion and/or reduce fluid reabsorption to augment impaired mucociliary clearance in CF represent a major line of therapeutics that address fundamental physiological deficits in CF, and are not restricted to specific CFTR mutations nor the uncertainties of gene transfer. A multifaceted approach via collaborations between scientists, industry, the CF Foundation, and the Therapeutic Development Network, utilising different therapeutic targets, has been pursued to maximise the chance of success. These alternative ASL restoring therapeutics can be grouped into those that promote increases in ASL volume and mucus hydration via osmotic mechanisms (eg, hypertonic saline; Bronchitol, Pharmaxis, Frenchs Forest, Australia), by reducing fluid reabsorption from the airway lumen through inhibition of the epithelial sodium channel (eg, GS9411, Gilead Sciences, Foster City, California, USA) or by increasing chloride secretion via various non-CFTR pathways (eg, denufosol, Inspire Pharmceuticals, Raleigh, North Carolina, USA; Moli 1901, Lantibio, Chapel Hill, North Carolina, USA; SPI-881, Sucampo, Bethesda, Maryland, USA). Within the osmotic hydrator class of therapies, hypertonic saline is already available,39 ,40 and successful completion of phase III trials of dry-powder mannitol in the USA and abroad led to the recent approval for use in Australia and the EU, whereas FDA review is presently underway in the USA.41–43 Addressing abnormal CF mucus, marked by the approval of recombinant human DNAse (Pulmozyme, Genentech, San Francisco, California, USA),44 has also had renewed interest, and is likely to lead to new lines of investigation supported by the network.
The concept of targeting sodium hyperabsorption, and thereby improving ASL hydration, first emerged more than three decades ago.45 ,46 The earliest attempts at manipulating sodium transport in the airways of patients with CF utilised inhaled amiloride.47–49 Treatment with amiloride was ultimately unsuccessful, likely due to inadequate potency and duration of action. Since then, newer agents with improved pharmacokinetic and pharmacodynamic properties have been developed, and early phase trials have been conducted to test their safety.50 Additional research with the latest molecules should be available soon for testing within the CF-TDN.
Stimulation of chloride secretion in the absence of CFTR can be accomplished in vitro and in vivo using agents that stimulate alternative chloride channels, including TMEM16a and others activated by calcium.51 Testing of an orally dosed prostone (cobiprostone, Sucampo) was conducted by the CF-TDN nearly 10 years ago. Although this study did not demonstrate a clear chloride secretory response, a related compound was eventually approved as a chloride secretagogue for the treatment of chronic constipation, and this study along with other early efforts informed the network on the use of NPD and other techniques now commonly used in CF-TDN network studies.25 ,52 Moli1901 (Lantibio) showed early promise as a chloride transport activator in US and European studies,53 but has not been pursued further due to formulation issues. Denufosol (Inspire Pharmaceuticals), a P2Y2 agonist designed to restore chloride transport and increase mucociliary clearance, also yielded promising results in early phase studies,54 ,55 but ultimately failed after the conduct of two phase III trials.56 ,57 Reflection on lessons learned and research into the potential cause of this failure has been undertaken and will educate the search for additional ASL modifying agents.
Anti-infectives
Chronic bacterial infections of the CF airways with resistant organisms, including multidrug resistant (MDR) Pseudomonas aeruginosa and other intrinsically resistant organisms such as Burkholderia cepacia complex, Stenotrophomonas maltophilia and Achromobacter species remain a major therapeutic challenge in CF.58 For example, the current rate of MDR P aeruginosa among patients with CF is 9.8%.59 Because of the severity and relentless nature of CF airway infections and the need to develop new treatment approaches to alleviate the development of resistance, an important focus of TDN-supported studies has been in the area of anti-infectives. Unfortunately, few new antimicrobials directed against P aeruginosa have been developed in the past two decades. Thus, recent anti-infective strategies have used currently available antimicrobials in new ways. In addition, novel approaches to treatment have been developed, including suppression of chronic infections and early therapy to eradicate infections prior to irreversible infection.
A sentinel agent in the development of new antimicrobial strategies for treatment of P aeruginosa in CF was tobramycin inhalation solution (TOBI, Novartis Pharmaceuticals, East Hanover, New Jersey, USA). Inhalation of antibiotics allows high concentrations of active drug at the site of infection, while minimising side effects. Inhaled tobramycin was tested in two trials including a total of 520 subjects over the age of 6 years. The trials demonstrated improvement in lung function and decrease in bacterial colony counts, and supported the drug's FDA approval in 1994. Although early in the formation of the network, the concept of inhaled suppressive antibiotics and the actual study design of the inhaled tobramycin trials have served as a model for other antimicrobial agents tested by TDCs. Aztreonam lysinate for inhalation (AZLI, Cayston, Gilead Sciences) was initially developed through support of the CFF and the clinical trials were performed through the TDN. Since its approval by the FDA, AZLI has been rapidly used by the CF population, demonstrating the intense need for new antimicrobial therapies. To extend efficacy to alternative organisms, a phase IV trial of AZLI for patients with CF and B cepacia complex infections was recently completed and Gilead is conducting an ongoing phase IV CFF Patient Registry-based study of antibiotic resistance. Other agents that have been formulated for airway administration and are currently in the CF Foundation Therapeutics Pipeline include tobramycin inhaled powder (TOBI Podhaler, Novartis Pharmaceuticals), inhaled levofloxacin (Aeroquin Axcan Pharmaceuticals, San Diego, California, USA), and inhaled liposomal amikacin (Arikace, Insmed, Monmouth Junction, New Jersey, USA), all of which are in phase III trials and offer improved convenience, delivery, and durability. Similarly, ciprofloxacin inhaled powder (Bayer Schering Pharma AG, Berlin-Wedding, Germany), has recently completed a phase II trial in CF, but has not yet begun phase III testing.
Another example of using currently available antimicrobials is the novel use of azithromycin in the management of CF airway infections. Although active in vitro against Staphylococcus aureus and Haemophilus influenzae, azithromycin is not considered active against the most typical gram-negative CF pathogens. However, studies developed through Cystic Fibrosis Foundation Therapeutics have demonstrated clinical response to three times a week administration of azithromycin. This was initially shown in a large clinical trial in patients with CF and P aeruginosa infection who were over the age of 6 and subsequently confirmed in younger children and those uninfected with P aeruginosa.60 ,61 The exact mechanism of action has not been demonstrated but patients responded with an improvement in FEV1 and a decreased frequency of pulmonary exacerbations.
Studies within the TDN have examined the role of early antibiotic therapy in eradicating early infection and delaying the establishment of chronic infections in young children.62–64 The Early Pseudomonas Infection Control (EPIC) trial was a multicentre randomised trial comparing one-drug or two-drug therapy (TOBI with or without oral ciprofloxacin) with either cycled or culture-dependent administration to eradicate early P aeruginosa infection. It demonstrated equal efficacy and safety in all arms without emergence of multiple antibiotic resistant organisms.65 ,66
A new antimicrobial soon to begin phase II testing is KB001 (KaloBios Pharmaceuticals, South San Francisco, California, USA), a monoclonal antibody fragment directed against the PcrV protein of the type 3 secretion system of P aeruginosa.67 Other antimicrobial therapies on the horizon include trials for the management of methicillin-resistant S aureus and non-tuberculous mycobacteria, and intravenous administration of gallium, an agent that is not a conventional antibiotic but has antimicrobial properties and is currently used by radiologists for nuclear medicine scans.
Anti-inflammatory therapies
Neutrophil-dominated airway inflammation is a hallmark of CF lung disease (see previous reviews68–71); thus, evaluating drugs that target inflammation is a major emphasis of the network. Corticosteroids and high-dose ibuprofen, each broad spectrum inhibitors of pro-inflammatory signalling, were two of the first anti-inflammatory drugs studied in CF. Both demonstrated clinical benefit,72–74 but side effects and other considerations have limited their use.75–77 However, the evidence that these anti-inflammatory agents can slow the progression of CF lung disease, particularly in children, suggests that strategies to modulate lung inflammation can be beneficial.
The CF-TDN has devoted significant resources to evaluating anti-inflammatory treatments. Among those successful include azithromycin, hydroxychloroquine which has been shown to reduce pulmonary exacerbations and improve lung function in patients with CF, without significantly affecting lower airway bacterial density60 ,61 (see Anti-infectives section). The evidence suggests that azithromycin may act as an anti-inflammatory or immunomodulatory agent, which may also explain its efficacy in chronic obstructive pulmonary disease.78 In a trial of azithromycin in patients with CF chronically infected with P aeruginosa, there was a modest reduction in sputum neutrophil elastase activity in favour of the azithromycin group.61 In a subsequent clinical trial in patients with CF, 6–18 years of age and uninfected with P aeruginosa, azithromycin significantly reduced circulating neutrophil counts and systemic markers of inflammation, including C-reactive protein, serum amyloid A and calprotectin.79
Several CF-TDN-supported studies evaluating more targeted anti-inflammatory agents have been largely unsuccessful, and a few were associated with detrimental side effects. A notable example of the latter was a trial testing the leukotriene B4 receptor antagonist amelubant (Boehringer Ingelheim, Bracknell, UK) in patients with CF and mild to moderate lung disease.80 The study was terminated early due to a significant increase in the frequency of pulmonary exacerbations in adults receiving amelubant. This trial gave the CF community pause, providing a potent reminder that modulating inflammation is not without risks. Based on preclinical models and associated clinical data,81 it was hypothesised that treatment with interferon γ (IFNγ), a pleiotropic cytokine with immunomodulatory and antimicrobial activities, would be beneficial in CF. However, a clinical trial with inhaled IFN-γ1b (Actimmune, InterMune, Brisbane, California, USA) did not significantly improve lung function, sputum bacterial density or sputum biomarkers of inflammation.82 The CF-TDN network also sponsored studies of ‘low-hanging fruit’ anti-inflammatory compounds (ie, available medications used for other inflammatory disorders), including the 3-hydroxy-3-methyl coenzyme A reductase inhibitor simvastatin, the peroxisome proliferator-activated receptor γ agonist pioglitazone, and the immunosuppressants methotrexate and hydroxychloroquine. Data from these small, short studies were largely negative, while methotrexate was associated with an intolerable adverse event profile.83–86 These compounds are no longer being actively investigated, and indicate the complexities of a targeted anti-inflammatory approach in CF lung disease. Additionally, recent 6-month antioxidant trials of inhaled glutathione and oral N-acetylcysteine, a glutathione prodrug, did not demonstrate positive effects either on inflammation, oxidative stress, or clinical outcomes in patients with CF.87 ,88
Despite these disappointments, there are several ongoing studies of potential therapeutic agents which address airway inflammation. Numerous studies have demonstrated an excess burden of neutrophil-derived proteases in the CF airway and provide justification for interventions that neutralise free neutrophil elastase activity or augment local antiprotease levels. There has been renewed interest by the pharmaceutical industry to develop α1-antitrypsin therapies in CF and clinical trials using more potent and efficacious formulations are once again being planned. Antioxidant deficiencies remain a viable therapeutic target in CF. A trial of a reformulated oral multivitamin supplement (AquADEKs), enriched with several non-enzymatic antioxidants including β-carotene, mixed carotenoids, coenzyme Q10, mixed tocopherols and selenium, will evaluate the effects of this antioxidant ‘cocktail’ on antioxidant levels and markers of oxidative stress. Other novel approaches include the phosphodiesterase inhibitor sildenafil on airway inflammation and the effects of sulforaphane (found in broccoli sprouts) on Nrf2 activation. Finally, a crucial question in the new era of CFTR modulation is whether improving CFTR activity will have a detectable impact on airway inflammation, a question addressed in the GOAL study (see CFTR modulation section). The breadth of these studies using the CF-TDN network suggest targeting inflammation in CF remains an attractive therapeutic approach, but optimising anti-inflammatory effects while minimising any detrimental impact on host defence remains a key challenge.
Nutritional therapies
Good nutrition is vital in CF,89 thus the TDN has investigated interventions to support normal growth. Almost 90% of patients with CF are pancreatic insufficient90 and need life-long treatment with pancreatic enzyme replacement therapy (PERT) to prevent malnutrition. Because some pancreatic enzymes products were available before passage of the 1937 Pure Food and Drug Act, drug companies had been allowed to manufacture and sell PERT without prior FDA approval.91 In 1994 case reports of fibrosing colonopathy (FC) began to appear; a subsequent analysis of CF Foundation Patient Registry data showed a strong correlation between dose of PERT and the appearance of FC.92 Concerns regarding PERT overuse, and other issues (eg, enteric coating and high-dose capsules) were raised as potential contributing factors. As a result of these and other problems, in 2004 the FDA issued notice that they would require a new drug application for PERT with a deadline of April 2008,93 which was subsequently extended to 2010. The CF-TDN played a central role in developing study protocols and implementing studies of the efficacy of PERT in this short but urgent time window. Based on these efforts, the FDA has approved five oral delayed-release PERTs, Creon (Creon Abbott Laboratories. Abbott Park, IL, USA),94 Zenpep (Zenpep Aptalis Pharma US, Inc., Birmingham, AL, USA),95 ,96 Pancreaze (Pancreaze Janssen Pharmaceuticals, Titusville, NJ, USA),97 ,98 Pertzye (Pertzye Digestive Care, Inc., Bethlehem, PA, USA) 99 and Ultresa (Aptalis Pharma US, Inc., Birmingham, AL, USA).
A longitudinal observational study of nutritional status in infants (BONUS) is underway, cosponsored by the National Institute of Health. With newborn screening for CF available in all 50 states as of 2010, PERT is being used in a larger number of infants, yet dose response has never been defined. A nested sub-study of BONUS will explore the efficacy of higher and lower doses of PERT in infants.
All current PERTs are biological products derived from hog pancreas. Although current FDA-approved products are no longer overfilled, have improved stability and are free of enveloped viruses, non-enveloped viruses cannot be removed from the drug substance without loss of efficacy. The CF-TDN has participated in studies of liprotamase, a novel formulation of recombinant bacterial lipase, fungal protease and amorphous amylase that is a non-biological PERT.100–103 The crystalline nature of some of its components confers stability in an acid environment and makes a liquid preparation possible, providing a potential advantage.
Progression of CF lung disease is linked to oxidative stress.104 Pancreatic insufficiency and a diminished bile acid pool cause malabsorption of important antioxidants including fat-soluble vitamins (vitamins A, D, E and K), carotenoids, tocopherols, selenium and coenzyme Q10.105 A number of studies have shown that in patients with CF, supplementation can correct many micronutrient deficiencies, although the evidence of clinical benefit has been hampered by inconsistent study design.106 The CF-TDN participated in developing an oral antioxidant-enriched multivitamin supplement that safely increased systemic antioxidant levels107 and continues efforts to explore the benefits of this nutritional approach to modify CF disease.
Biomarkers
Since the inception of the CF-TDN, the development of biomarkers to enhance CF clinical trial planning and conduct has been at the forefront of network activity since alternatives to hard clinical endpoints such as mortality and hospitalisation are not viable endpoints for phase II and many phase III programmes.4 Biomarkers are ‘a characteristic that is objectively measured and evaluated as an indicator of a normal biologic process, pathogenic process, or pharmacologic response to a therapeutic intervention’.108 As such, CF-TDN-supported research has concentrated on the testing and validation of new biomarkers, linking changes in biomarkers to established surrogate or clinical outcome measures, and refining biomarker assessment techniques largely in the context of drug development. In addition, the acquisition of high-quality longitudinal biospecimens linked to clinical data has been incorporated into several CF-TDN clinical trials, leading to the development of rich specimen banks for biomarker discovery. For example, these biobanks have been leveraged by the CF research community to help define relationships between sputum inflammatory markers (eg, interleukin-8, human neutrophil elastase) and clinical outcome measures (eg, lung function),8 ,109 and to relate macrolide therapy to anti-inflammatory activity.79 The capacity to interrogate samples obtained from multicentre studies that have been collected with common standard operating procedures also increases the probability that results can be successfully recapitulated in the clinic.
With the development of CFTR modulators, there has been a need to develop and refine biomarkers related to augmented CFTR function. From these efforts, sweat chloride has emerged as a robust, standardised and validated method to monitor the bioactivity of CFTR potentiators and correctors.6 ,17 ,19 ,20 ,110 NPD has also performed reasonably well as a CFTR biomarker,111–113 successfully detecting ivacaftor activity in patients with G551D CF. For unclear reasons, neither CFTR biomarker has correlated with clinical outcomes (eg, lung function, patient-reported outcomes (PROs), weight) on an individual patient basis. Ongoing CF-TDN trials are underway to better understand this gap; one example is the GOAL study, which includes the evaluation of functional biomarkers (see CFTR modulation section), banking of serum, sputum, and urine linked to clinical data, and may help future biomarker discovery.
A critical barrier that can potentially limit the extension of new therapies to the treatment of infants and toddlers with CF (and other patients with very mild obstructive disease) is the relative lack of biomarkers sensitive to small changes in these populations. It is a widely held opinion that the greatest benefits conferred by many CF therapies would be expected in the healthiest patients, prior to the development of irreversible lung injury. As one example of success in this area, the CF-TDN developed standard operating procedures for the conduct of infant pulmonary function tests for multicentre performance and analysis.114 These efforts were used to demonstrate improved lung function (change in FEV in 0.5 s) in infants and toddlers with CF treated with 7% hypertonic saline compared with normal saline, despite no detectable difference in pulmonary exacerbation rates between the two treatment groups.115 Other CF-TDN studies in progress are geared towards accelerating drug development in the infant population, including the Baby Observational and Nutritional Study which also includes a biospecimen collection programme (see also above). Finally, TDCs have participated in non-invasive techniques to monitor mild lung disease and responses to intervention, including CT imaging55 ,116–119 and Lung Clearance Index measurements in ivacaftor-treated patients with G551D CF. Further studies that link sensitive disease biomarkers to clinical endpoints in the setting of new and established therapies could help accelerate the development path required to bring therapies to the healthiest patients with CF.
The TDN has played an instrumental role in advancing CF PROs and helping define their role in drug development. This work was particularly instrumental during the development of AZLI.120–122 PROs have also served as primary and secondary endpoints in pivotal trials involving CF therapeutics; establishing the minimal clinically important difference for the Respiratory Domain of the CF Questionnaire-Revised (CFQ-R) was critical to this effort.123 This work has paved the way for the incorporation of the CFQ-R into clinical trials of many new CF treatment strategies.6 ,17 ,19 ,20 ,124 ,125 Future studies are planned to further refine CF PROs, including the development of PROs for non-pulmonary disease manifestations, correlation of PRO results with other biomarkers, and the capture of information more frequently (eg, daily diaries compared with 2-week recall that is a limitation of the current CFQ-R).
Future directions for the network
As novel therapies emerge and evolve, clinical trials infrastructures must respond to the expanding need for studies. The CF-TDN and its partner network, the European Society for Cystic Fibrosis (ESCF) Clinical Trials Network, must maintain high-quality study conduct, which will be optimised by providing the structure and tools for improvement through web-based, QI programmes. These programmes are available to all sites with the requirement that they participate in an assessment–prioritisation–implementation–reflection cycle and report their results annually. A competitive application process will continue to sustain an engaged network of participating sites that allows new sites to enter the network when they demonstrate interest and capability, and permits adjusting network size to meet enrolment projections for studies linked to the research pipeline.
The CF-TDN will continue to work cooperatively with the ESCF Clinical Trials Network and the Canadian and Australian CF research communities to facilitate the conduct of uniform protocols across multiple countries, to enrol larger sample sizes in a shorter time period, and permit sufficient recruitment of subsets of patients with unique characteristics, such as age, genotype, infecting organism or comorbidities. Collaboration across networks also exists for combined protocol reviews, development of industry and investigator initiated studies, and working groups developing consensus around novel outcome measures and endpoint definitions. Similarly, the network has made a consistent effort to learn from challenges experienced, and address these areas in the future; some important examples are noted in table 3.
Lessons learned by the TDN and changes implemented to address them
The development of novel or improved outcome measures remains a major focus for CF-TDN working groups; in particular, assessing outcome measures applicable for infants and young children as new therapies become available and decisions regarding efficacy in younger populations must be made, for example.126 The existence of the CF Patient Registry enhances the conduct of clinical research in many ways, and will be a pivotal tool for implementation of comparative effectiveness research (CER) in CF. The tool has already been used in long-term observational studies conducted through the CF-TDN.127 As this capability grows, it is conceivable that it will allow the expansion of future CER and observational studies to all CF care centres that participate in the registry, maximising investigative power. Combined with the robust pipeline of therapeutics, the network is well poised to make continued contributions to the rapid evolution of CF care.
Acknowledgments
The authors acknowledge grant support including CLANCY09Y0, CLANCY05Y2, and UL1 TR000165 (SMR), BOROWI03CS0 (DSB), RETSCH09Y0 and UL1TR000083 (GRB and SHD), AMIN09YO (JPC), RAMSEY03Y0, 1P30DK089507 and UL1TR000423-06 (JLB and BMR) and SAGEL07B0 (SDS).
References
Footnotes
-
Contributors All named authors wrote and edited the manuscript. SMR takes final responsibility for the content. No ghost writers were affiliated with the work.
-
Funding CFF and NIH.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves