Article Text

Abstract
Background Culture-independent analysis of the respiratory secretions of people with cystic fibrosis (CF) has identified many bacterial species not previously detected using culture in this context. However, little is known about their clinical significance or persistence in CF airways.
Methods The authors characterised the viable bacterial communities in the sputum collected from 14 patients at monthly intervals over 1 year using a molecular community profiling technique—terminal restriction fragment length polymorphism. Clinical characteristics were also collected, including lung function and medications. Ecological community measures were determined for each sample. Microbial community change over time within subjects was defined using ecological analytical tools, and these measures were compared between subjects and to clinical features.
Results Bacterial communities were stable within subjects over time but varied between subjects, despite similarities in clinical course. Antibiotic therapy temporarily perturbed these communities which generally returned to pretreatment configurations within 1 month. Species usually considered CF pathogens and those not previously regarded as such exhibited similar patterns of persistence. Less diverse sputum bacterial communities were correlated to lung disease severity and relative abundance of Pseudomonas aeruginosa.
Conclusion Whilst not true in all cases, the microbial communities that chronically infect the airways of patients with CF can vary little over a year despite antibiotic perturbation. The species present tended to vary more between than within subjects, suggesting that each CF airway infection is unique, with relatively stable and resilient bacterial communities. The inverse relationship between community richness and disease severity is similar to findings reported in other mucosal infections.
- Bacterial infection
- cystic fibrosis
- respiratory infection
- lung cancer
- bronchiectasis
- COPD mechanisms
- opportunist lung infections
- pneumonia
Statistics from Altmetric.com
- Bacterial infection
- cystic fibrosis
- respiratory infection
- lung cancer
- bronchiectasis
- COPD mechanisms
- opportunist lung infections
- pneumonia
Key messages
What is the key question?
-
To what degree do the bacterial species present in cystic fibrosis (CF) sputum represent chronic colonisation of the lower airways?
What is the bottom line?
-
The CF bacterial community, including species only recently reported, as well as known pathogens such as Pseudomonas aeruginosa, is in many instances stable and resilient over extended periods.
Why read on?
-
These findings provide a basis for understanding the clinical importance of the bacterial community in disease progression, and a context for studies that assess the impact of antimicrobial therapy.
Introduction
Whilst advances in longevity continue, respiratory failure remains the leading cause of death in cystic fibrosis (CF). The lung damage that leads to respiratory failure results from the chronic bacterial infection and host immune response that is typical by adulthood, if not before. Infection by certain bacterial species has been associated with poorer clinical outcomes.1 ,2 These species have been detected previously using selective culture-based approaches that are inherently biased. More recent studies are increasingly characterising the bacteria present in the CF airways using culture-independent approaches that avoid these biases.3–6 In this way, much greater bacterial diversity has been found typically in these airway secretions than previously appreciated. It is also important to note that bacterial species, many of them anaerobes, identified using culture-independent methods are frequently of abundances comparable to species routinely cultivated and treated as pathogens in the CF airway.6–8
From the earliest of these culture-independent studies, it has been postulated that species present in the lower airways represent an interacting community,9 implying roles for ‘non-traditional’ species in CF lung disease pathogenesis and treatment response. However, support for such a community model requires determination of the persistence of the various community members, the community response to perturbations, for example antibiotics, and its relationship with disease severity. Some species may remain in the lower airways for relatively short periods, while others may establish chronic infections. Infection over longer periods may also be more likely to adversely impact upon respiratory health. In support of these concepts, CF airways communities have been suggested to contain both core and transient species,10 with community structure and membership related to age and lung disease severity.11 However, such studies are generally cross sectional or short term,12 and a longitudinal analysis of multiple subjects has not yet been performed.
Longitudinal analysis has followed how specific CF pathogens change with time during chronic airway infection.13 ,14 This approach must now be applied to the entire community. The first step is to define the variability of CF airway microbiota over time, during clinical changes, and with antibiotic treatment. Community constituency could change in a number of ways. At one extreme, the composition of the community may change frequently, with the community found in one specimen not resembling those in subsequent samples from the same subject. At the other extreme, the community may be so stable that the same species are recovered at any sampling point. CF airway disease is typified by periods of relative respiratory health punctuated by exacerbation; with augmented treatment, usually requirement for antibiotics. As such, we hypothesised that the varying treatment that a patient with CF receives over the course of a year, and the dynamic nature of respiratory and general health, would be associated with detectable changes in the bacterial community and a ‘highly variable’ composition. To test this hypothesis required the collection of samples from multiple subjects spanning an extended period.
Here, we report the culture-independent analysis of viable bacteria present in monthly samples from airways of 14 adults with CF over a year. These data were collected alongside detailed clinical metadata. A series of analytical tools tested the compositional stability of these communities over time.10 We also analysed the relationship between community structure and lung disease severity.
Materials and methods
Clinical samples
This observational study of 14 adult patients with CF over 12 months was undertaken with ethical approval from Southampton and South West Hampshire Research Ethics Committee (06/Q1704/26). Eligible subjects were aged 18 or over and had experienced at least three pulmonary exacerbations (CFPEs) requiring antibiotic treatment in the prior 12 months. Samples from these patients were previously used in an analysis of core and satellite bacterial taxa.10
Patient clinical details are summarised in supplementary table 1. The start of a CFPE was defined by the clinician's decision to initiate antibiotic therapy for deteriorating clinical status, broadly based on factors described previously.15 The end of CFPE was defined by the decision to cease antibiotic therapy due to stabilisation or improvement in signs and symptoms. A total of 39 episodes of CFPE were experienced by these patients over the year, ranging from 0 (patients 3 and 8) to 6 (patient 9) with a mean of 2.8, SD 1.4. Overall, 17 different antibiotic combinations were used with five ‘elective’ antibiotic courses given for reasons other than worsening respiratory symptoms.
One sputum sample was obtained from each patient at approximately monthly intervals. The majority of samples (65%) were collected at least 21 days prior to, or 21 days after cessation of, antibiotics for CFPE (supplementary table 2). All sputa were stored at 4°C immediately after expectoration, shipped at 4°C (in accordance with handling guidelines16) and stored at −80°C prior to processing, or cultured by the Health Protection Agency South East, UK following standard operating procedures.
Clinical data
Clinical measures (including forced expiratory volume in 1 s (FEV1) and forced expiratory volume in 6 s (FEV6) in litres, temperature, and patient reported outcome (PRO) scores) were recorded at sample collection. PRO scores were recorded using visual analogue scores for individual symptoms (‘breathlessness’, ‘cough severity’, ‘sputum production’ and ‘general well-being’) from 0 (no symptoms) to 100 (worst symptoms).
Culture-independent analysis
Exclusion of DNA from non-viable cells in sputa via cross-linking using propidium monoazide17 ,18 and subsequent nucleic acid extraction were performed as described previously.19 PCR and terminal restriction fragment length polymorphism (T-RFLP) profiling were carried out as previously described3 (see supplementary materials and methods).
Statistical analysis
Bacterial species richness (the number of bacterial species in a sample) was inferred from T-RF band number as previously described.20 Cumulative bacterial taxon richness (the total number of different bacterial taxa identified after each successive time point) was assessed using taxa–time relationships (TTRs); these describe how richness increases with the time over which the community is monitored20 ,21 (see supplementary materials and methods).
Distance–decay relationships (DDRs) of bacterial community similarity with time (temporal differences in days) were determined. DDRs describe how similarity in taxa composition between two communities varies with a measure of distance (here, the time between pairs of sample collections)22 (see supplementary materials and methods).
Principal component analysis (PCA), used to identify differences in community structures, was carried out with software developed in house, using the Python programming language with the Numpy package. The principal components were calculated using eigenvalue decomposition of mean-centred and auto-scaled data.
Results
Measures of community species constituency, and clinical data, were compared for sputa collected monthly over a year from 14 patients. Bacterial community measures assessed the changes in the relative abundance of species detected both within individuals over time, and between subjects.
Bacterial diversity
Bacterial species dynamics were determined using selective culture-based analysis and DNA-based profiling. Culture-based microbiological analysis (supplementary table 3) showed that Pseudomonas aeruginosa and Pseudomonas spp. were the most frequently reported (70.8% of samples). Routine culture also commonly identified ‘oral flora’ (56.0%). ‘Unidentified isolates’ (1.2%), ‘coliform’ (2.4%), ‘Stenotrophomonas maltophilia’ (0.6%), ‘methicillin-sensitive Staphylococcus aureus (MSSA)’ (10.7%), ‘Staphylococcus sp.’ (1.8%) and ‘Streptococcus Group F’ (0.6%) were also reported. No significant correlations were found between species detected by this method and clinical status, or in the coculture of separate species (Pearson correlations with Bonferroni corrections).
The second tool used to analyse bacterial species dynamics was the culture-independent technique, T-RFLP. T-RFLP provides measures of species constituency when based on 16S rRNA gene analysis. In the 168 samples, a total of 1158 T-RF bands were detected, representing 37 distinct T-RF lengths (each representing one or more species). The mean number of T-RF bands in profiles from individual patients over the study ranged from 2.6 (SD 2.4, n=12) (patient 12) to 12.7 (SD 1.6, n=12) (patient 10), with an overall mean of 6.9 (SD 3.9, n=168).
T-RFLP data were used to characterise which species were present over the study period in each subject. In the following sections, data for three patients are selected to represent the types of community dynamics observed; all other data are presented as supplementary information. These patients represented the highest (patient 2) and lowest (patient 4) relative bacterial community stability (Bray-Curtis similarity index), and the relative stability closest to the group mean (patient 13).
Community change over time
TTR analysis identified the cumulative number of species detected in each sample (figure 1 for patients 2, 4 and 13; remainder in supplementary figure 1). For the overall dataset, scaling component values (w) were observed to range from 0.05 (patient 6) to 0.38 (patient 12) (supplementary figure 1). This indicated that some patients acquired new species more frequently than others. No significant relationship was identified however between the number of species at the beginning of the study and the subsequent rate of new species acquisition (pairwise Pearson correlation across all patients, p=0.699, r2=0.5).

Species richness dynamics and taxa–time relationship. Species richness (black circles, black line) and the cumulative species richness (white circles, no line) are shown. Power regression lines were fitted to the cumulative species richness. Also given are the taxa–time relationship power law equation (S = cTw), and coefficients of determination (r2). All regression coefficients were significant (p<0.001).
The overall change in community composition over the 12-month sampling period was assessed using the inverse of the Bray-Curtis similarity index. This was applied to all possible pairs of samples from individual subjects and the mean of each subject's resulting values calculated for the year (n=11) (results expressed as % change; supplementary table 4). While the mean change in bacterial composition for the group overall was 35.5% (SD 18.2%, n=154 samples), low levels of change were seen in certain patients (eg, patients 2, 3 and 5 had 4.3%, 18.1% and 18.8% change respectively; supplementary table 4), with higher relative levels of change seen in other patients (eg, patients 4 and 14 had 70.1% and 64.3% change, respectively; supplementary table 4). Together, these results indicated that bacterial community variation is inherently different among patients with CF. No relationship was identified between mean richness of a patient's community and its variability over time (Pearson correlation, p=0.252, R=0.389).
To test whether similarity between samples was due solely to the time interval between sample collection, DDR were used (figure 2 and supplementary figure 2). A statistically significant distance-decay relationship was identified for two subjects (4 and 7, p=0.040 for both). For all other subjects, no relationship between community composition at two given sample points and time interval between them was found (supplementary table 5). This suggests that the community stability observed was not simply a function of sampling frequency.

Distance–decay relationships. The distance–decay of bacterial community similarity with time. Power regression lines were fitted, and power law regression equations (SBC = cTd) and coefficients of determination (r2) are given in each instance. Only two relationships were significant (p=0.040 for patients 4 and 7).
PCA was performed on community data from each subject (figure 3 and supplementary figure 3). While the communities in each patient varied over time, no trends within patients were observed consistently across the patient group as a whole. Further, samples collected during periods of exacerbation were not found consistently to be significantly different from non-exacerbation communities (Pearson correlations for community richness, p=0.216, R=0.501, structure (slope), p=0.056, R=0.765, and P aeruginosa relative abundance, p=0.023, R=0.648). PCA analysis on data from all subjects are shown in figure 4. When the two components that explain the highest levels of variance are examined, the sample points from individual patients cluster more tightly with each other than with communities from other subjects in a number of cases. The relatively small proportion of total variation represented by any individual component (14% for component 1) indicates that bacterial community differences between patients cannot be ascribed to differences in a small number of correlated factors, but rather are likely to represent many unrelated factors, each contributing a small amount of variance.

Principal components analysis of individual patients. Plots for community profiles by component 1 (PC1) and component 2 (PC2) are shown. The percentage of variance in the data explained by each component is indicated on the relevant axis. Sampling points are indicated with numbers 1–12, with samples collected during pulmonary exacerbations indicated by boxed numbers.
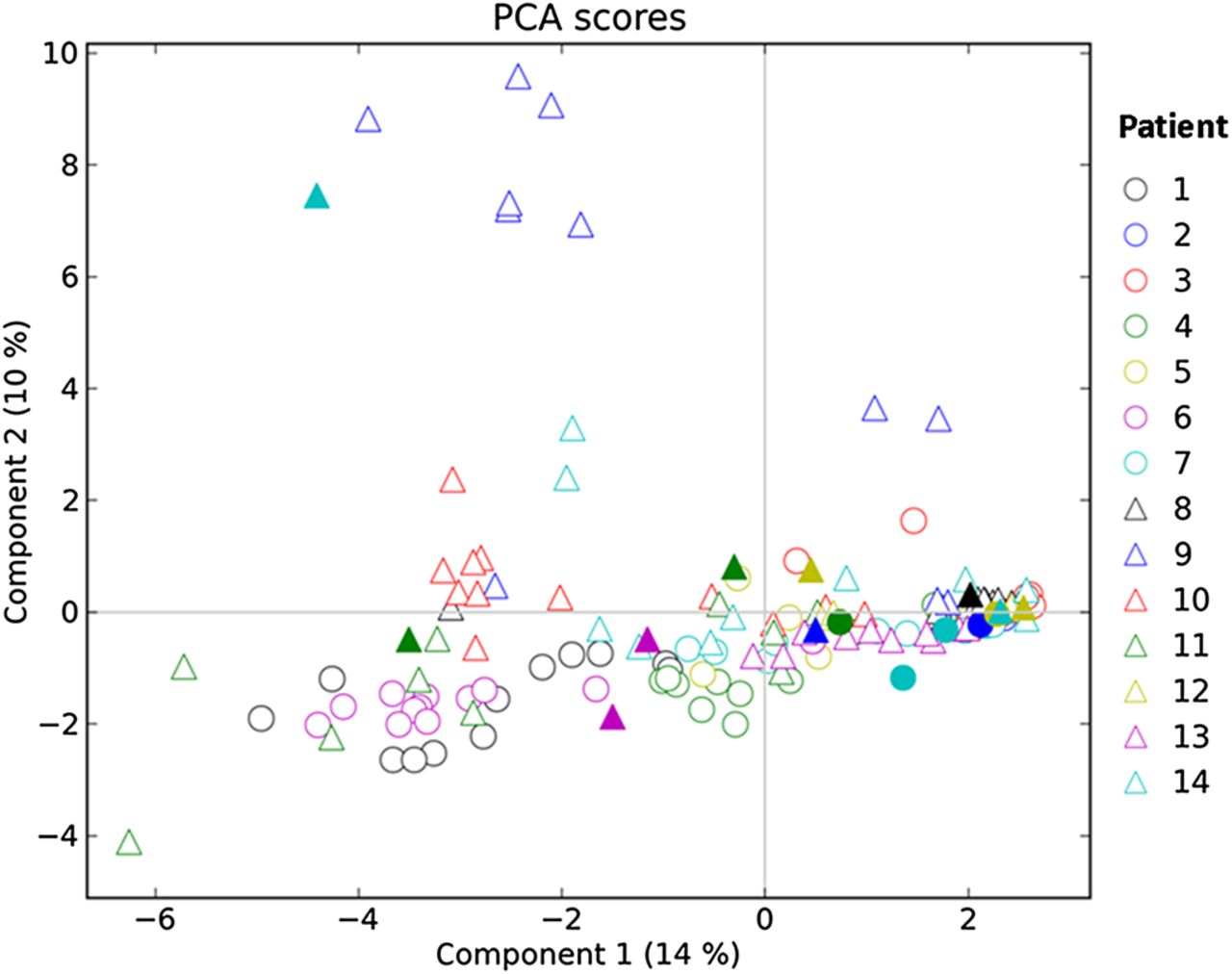
Principal components analysis (PCA) of all subjects. The variance explained by each component is shown in brackets. Solid shapes indicate samples collected during pulmonary exacerbations.
Clinical data
Figure 5 shows the variation in FEV1, PRO measures and antibiotics for patients 2, 4 and 13 respectively; data for all patients are in supplementary figure 4, with FEV6 data shown in supplementary figure 5. The FEV1 (% predicted) values ranged from 5.6% (patient 9) to 94.2% (patient 10), mean 49.1% (SD 17%, n=163). FEV6 (litres) values ranged from 0.56 (patient 2) to 3.85 (patient 10), mean 2.27 (SD 0.79, n=162). To reduce the effect of short-term lung function fluctuations, a weekly mean for FEV1 and FEV6 was also calculated (here, the mean was calculated for all measurements 3 days preceding and following each sampling point). PRO scores are also displayed as a sum of the respective values for the component symptom scores (breathlessness, sputum production, cough and general wellbeing).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forced expiratory volume in 1 s (FEV1), patient reported outcomes (PROs) and antibiotic treatment at sampling. Lung function FEV1 (litres) (black diamonds) is shown at each sampling point, and a calculated weekly mean FEV1 (black solid line). The individual patient reported outcome scores (breathlessness, sputum production, cough, general wellbeing) were summed for each sampling point and are shown as grey areas (the higher the score, the worse the symptoms). Pulmonary exacerbations with antibiotic treatments are indicated as black dashes at the bottom of the graph with the details for each pulmonary exacerbation (marked with an asterisk and the respective number) shown in supplementary table 1.
Apart from patients 1, 2 and 11, for whom a substantial decline was observed, lung function for the other patients was relatively stable over the year. PRO scores varied greatly between patients, however no clear relationship was observed in relation to lung function changes.
Correlating clinical and microbial datasets
Airway community characteristics were correlated with clinical characteristics. Subjects were separated into higher and lower halves based on FEV1, with these resulting groups examined for community characteristic differences. Five of the six patients with FEV1 above the median (patients 1, 6, 10, 11 and 14) shared three or more of the following characteristics: species number >7, a mean relative P aeruginosa abundance of <35%, a TTR value of <0.1, and a DDR value of <0.1 with no statistical significance. All of these patients had experienced at least three CFPEs, except patient 6 (two CFPEs), and showed a change in community composition over the year above the median (31.7%), except patient 6 (24.8%). In contrast, only one subject with a low mean FEV1 (patient 9) had four of these criteria.
The statistical significance of these relationships was tested through Pearson's coefficient correlation analyses (table 1). Three significant relationships were identified after the application of Bonferroni correction; higher PRO scores reported by women than men (p=0.002, R=0.758); a steeper rank abundance slope (the slope of the regression line fitted to species abundance ordered from highest to lowest abundance) with detection of low species number (p=0.002, R=−0.763) and a steeper rank abundance slope correlated with high P aeruginosa relative abundance (p=0.002, R=−0.763).
Summary of significant correlations*
In addition, pairwise comparisons between clinical and microbiological data were performed across all 168 samples independent of patient identity. Significant relationships after Bonferroni correction between FEV1 and FEV6 and higher species number per sample, steeper rank abundance slope, and lower P aeruginosa relative abundance were identified (table 1).
Discussion
This study defined the stability of communities over time in relation to host and clinical characteristics. In general, while some degree of overlap was observed, the airways of individuals with CF contained communities that differed from those in other subjects, often despite similar clinical courses. In contrast, subjects' bacterial communities changed little over the year, despite intervening respiratory exacerbation periods; the changes observed during antibiotic treatment did not persist, returning to approximate pretreatment structures within a month.
Of the previous studies that applied culture-independent methods to examine the microbiota of CF airways,3 ,5 ,6 ,23 ,24 two focused on the relationship with lung disease severity. In agreement with these studies,10 ,11 subjects with higher lung function in our study tended to have more diverse airway communities and vice versa. Furthermore, as also shown previously,10 ,11 ,23 P aeruginosa was associated with lower community richness and lower lung function.
The feature that distinguished this study from previous work was its longitudinal analysis of viable cells. Most earlier studies identified the bacteria in individual CF samples in cross-sectional analyses or, less often, in two or more samples taken over a short time period (eg, in treatment of exacerbation).8 ,12 Tunney et al 12 applied a combination of T-RFLP and culture to demonstrate that sputum communities changed little during and soon after antibiotic treatment. While this and other short-term studies were revealing, a longer-term study of multiple subjects was required to define long-term airway bacterial dynamics. We sought to distinguish transient from chronic infectious characteristics, as the latter are more likely to be associated with lung disease progression. While Sibley et al 6 previously analysed bacterial dynamics over a period comparable to that examined here, their study focused on a single individual, precluding generalisation and inter-subject comparison.
Further, we focused on only viable cells18 ,19 to ensure an accurate characterisation of community stability. This minimised the contribution of dead bacteria, for example, from antibiotic treatment or immune response. Thus, bacterial species consistently observed can be described with some confidence as being chronically present in the CF lower airways.
Given the many perturbations experienced by the airway microbiota in an adult patient (including antibiotic therapy, immune response, and cough clearance), we hypothesised that each subject's bacterial community would vary greatly in membership over time. Cross-sectional analyses of bacterial communities in CF airways have reported a wide range of bacteria.10 ,11 Such variation could be explained by high species turnover. Surprisingly though, we found CF lung bacterial communities to be, to a large extent, stable over a year.
In contrast, the composition of airway communities differed among patients. Interestingly, this was reminiscent of findings from other mucosal microbial communities, for example, the gastrointestinal tract.25 The reasons for this stability within patients, and divergence between patients, are unknown; however, disease severity and its correlate, treatment history, may play selective roles. We also observed that CF airway bacterial communities were resilient. While antibiotic therapy can have a significant short-term impact on CF lung communities,8 ,12 our data indicate that these communities approach pretreatment configurations rapidly.
Our results also extend the previous finding that CF lung community richness is inversely correlated with lung disease severity.10 Neither community richness nor lung function changed significantly within subjects during the study. As such, we could not define the contribution of individual species, or specific communities, to lung disease pathogenesis. However, we did find an inverse relationship between community richness and lung function, similar to previous cross-sectional studies.11 This is reminiscent of relationships found for other mucosal microbial communities.26 ,27 In this regard, while no causality can or should be inferred, the current results may indicate that CF lung health is highest in the presence of a diverse microbiota. Alternatively, repeated antibiotic exposure, or the toxic products of pathogens such as P aeruginosa, could be responsible for community simplification. Antibiotic treatment could not be controlled for in this study. As such, further investigation of the relationships observed between community richness and disease severity is now warranted.
In summary, we found that the species that constitute CF airway communities are present for prolonged periods, and that communities can change little over a year, despite antibiotic treatment. This stability indicates that much of the wider bacterial diversity seen in airway samples does not represent short-term colonisation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online Materials
- Data supplement 1 - Online Supplementary Tables
Footnotes
Linked article YES
-
Funding This work was supported by the Anna Trust (grant number KB2008) and the Seattle Children's Hospital Center for Clinical and Translational Research.
-
Competing interests None.
-
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
-
Ethics approval Ethics approval was provided by Southampton and South West Hampshire Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves