Article Text

Abstract
Background VX-809, a cystic fibrosis transmembrane conductance regulator (CFTR) modulator, has been shown to increase the cell surface density of functional F508del-CFTR in vitro.
Methods A randomised, double-blind, placebo-controlled study evaluated the safety, tolerability and pharmacodynamics of VX-809 in adult patients with cystic fibrosis (n=89) who were homozygous for the F508del-CFTR mutation. Subjects were randomised to one of four VX-809 28 day dose groups (25, 50, 100 and 200 mg) or matching placebo.
Results The type and incidence of adverse events were similar among VX-809- and placebo-treated subjects. Respiratory events were the most commonly reported and led to discontinuation by one subject in each active treatment arm. Pharmacokinetic data supported a once-daily oral dosing regimen. Pharmacodynamic data suggested that VX-809 improved CFTR function in at least one organ (sweat gland). VX-809 reduced elevated sweat chloride values in a dose-dependent manner (p=0.0013) that was statistically significant in the 100 and 200 mg dose groups. There was no statistically significant improvement in CFTR function in the nasal epithelium as measured by nasal potential difference, nor were there statistically significant changes in lung function or patient-reported outcomes. No maturation of immature F508del-CFTR was detected in the subgroup that provided rectal biopsy specimens.
Conclusions In this study, VX-809 had a similar adverse event profile to placebo for 28 days in F508del-CFTR homozygous patients, and demonstrated biological activity with positive impact on CFTR function in the sweat gland. Additional data are needed to determine how improvements detected in CFTR function secondary to VX-809 in the sweat gland relate to those measurable in the respiratory tract and to long-term measures of clinical benefit.
Clinical trial number NCT00865904
- Cystic fibrosis
- ion transport
- sweat chloride
- protein folding disorders
- paediatric lung disaese
- respiratory infection
- bronchiectasis
- cystic fibrosis
- atypical mycobacterial infection
Statistics from Altmetric.com
- Cystic fibrosis
- ion transport
- sweat chloride
- protein folding disorders
- paediatric lung disaese
- respiratory infection
- bronchiectasis
- cystic fibrosis
- atypical mycobacterial infection
Key messages
F508del-CFTR is the most common cystic fibrosis-causing mutation. Restoring function to F508del-CFTR offers a novel treatment strategy for the majority of patients with cystic fibrosis. VX-809 had an adverse event profile similar to placebo in F508del-CFTR homozygous CF patients, and reduced sweat chloride values in a dose dependent manner. The findings indicate that F508del-CFTR is a viable target for drug development.
Introduction
Cystic fibrosis (CF) is the most common autosomal recessive lethal genetic disease in the Caucasian population, with an incidence of 1:3500 births in the USA. The most common cause of morbidity and mortality is lung disease, which is characterised by infection, inflammation and airway damage that leads to respiratory failure.1
CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene,2 3 which encodes the CFTR protein. CFTR is a member of the ATP-binding cassette protein family and functions as a chloride ion (Cl−) channel and a key regulator of salt and water transport across a variety of epithelia.4–8 The CFTR gene has >16009 reported disease-associated mutations, with F508del-CFTR being the most common. F508del-CFTR results from a 3 bp deletion that leads to the omission of phenylalanine at position 508 of the full-length protein.10 The resulting F508del protein product is unstable and susceptible to rapid degradation in the 26S proteosome, with little if any F508del-CFTR at the plasma membrane.11 12 The F508del-CFTR mutation is found in the majority of patients with CF,13 and therefore the consequences of this mutation on the CFTR protein are important to address in therapeutic development for CF.
CFTR ‘correctors’ aim to increase the cell surface density of functional CFTR protein, resulting in improved chloride transport and decreased sodium reabsorption.14–16 VX-809 restores F508del-CFTR processing and plasma membrane localisation in primary human bronchial epithelial (HBE) airway cells isolated from patients homozygous for the F508del-CFTR mutation, achieving ∼15% of wild-type CFTR levels as measured by the amount of chloride channel function and the quantity of fully mature, C-Band CFTR.17
In this report, we describe the safety, tolerability, pharmacodynamics and pharmacokinetics (PK) of escalating doses of VX-809 in patients with CF homozygous for the F508del-CFTR mutation compared with placebo.
Methods
This study was a randomised, double-blind, placebo-controlled, multiple-dose, multicentre, phase IIa study. Institutional Review Board approvals and informed consents were obtained for all study subjects.
Subjects
Subjects enrolled in the study had a confirmed diagnosis of CF, accompanied by a sweat chloride value of ≥60 mmol/l. All subjects were ≥18 years of age, and were required to have the F508del-CFTR mutation on both alleles. At screening, subjects were required to have a forced expiratory volume in 1 s (FEV1) of at least 40% of predicted normal for age, gender and height (Knudson standards).18
Study design
Subjects were enrolled into two cohorts, group A and group B. Group A subjects were randomised to receive VX-809 once daily at doses of 25 mg or 50 mg, or placebo in a 2:2:1 randomisation ratio. After 15 group A subjects completed 28 days of treatment, a safety review was conducted by an independent Data Monitoring Committee. Enrolment in group B began following the Data Monitoring Committee review. In group B, subjects were randomised to receive VX-809 at doses of 100 mg or 200 mg, or placebo in a 2:2:1 ratio for 28 days. All study sites, the patients, and the sponsor remained blinded to treatment assignment throughout the study.
End points
The primary objective, evaluation of safety and tolerability of VX-809, was assessed by adverse events (AEs), haematology, clinical chemistry, urinalysis, ECG, vital signs and physical examinations. Secondary objectives included evaluation of the pharmacodynamic impact of VX-809 on CFTR function. Measures of CFTR activity included sweat chloride and nasal potential difference (NPD).19 The latter was considered optional. Other secondary end points included spirometry to measure pulmonary function (ie, FEV1, forced expiratory flow 25–75% (FEF25–75%) and forced vital capacity (FVC)) and the CF Questionnaire-Revised (CFQ-R), a disease-specific patient-reported outcome.20 The minimal clinically important difference in respiratory domain (MCID) is improvement ≥4.21 The details of CFTR biomarkers and pharmacokinetic analysis are included in the Supplemental Methods section.
Statistical analyses
Safety data were analysed primarily using descriptive statistics, and efficacy data were analysed primarily using analysis of covariance (ANCOVA) models. The planned total sample size of 90 subjects provided a study power of >97% to detect a 20 mmol/l reduction in sweat chloride concentration from baseline, and a probability of 99% to observe at least 1 adverse event in the study. All subjects who received at least one dose of study drug were included in the analyses.
Results
Subjects
A total of 109 adult subjects with CF who were homozygous for the F508del-CFTR mutation (based on screening medical history) were screened and 89 were randomised to one of four VX-809 dose groups (25 mg (n=18), 50 mg (n=18), 100 mg (n=17), 200 mg (n=19)) or to placebo (n=17). Confirmatory genotyping identified one subject randomised to the 50 mg VX-809 group who was heterozygous for F508del-CFTR despite a medical history indicating homozygosity. Safety and efficacy data obtained from this subject were included in the final analysis. The identity of this subject's non-F508del allele was not available (based on parameters of the informed consent document for that study site). Baseline characteristics are provided in table 1. Median sweat chloride values and FEV1 percentage predicted were similar between the different groups and consistent with the values reported in the literature for patients homozygous for the F508del-CFTR mutation. The baseline median sweat chloride was 103.5 mmol/l and the median baseline FEV1 was 71% predicted. The study groups were well matched except for trends towards less severe lung disease in the placebo and 25 mg dose groups.
Baseline demographic and clinical characteristics of the study subjects
Safety and AE profile
The incidence of AEs was similar between dose groups (table 2). Respiratory events were the most commonly reported type of AE, with cough occurring in 46% of VX-809-treated subjects and 41% of placebo-treated subjects. There was no difference in the incidence of physician-diagnosed pulmonary exacerbations between VX-809- and placebo-treated subjects (17% of VX-809 subjects compared with 12% of placebo subjects; p=0.62). AEs that occurred in more than one subject in any VX-809 treatment arm are included in table 2. Eight AEs were considered severe, including fatigue, sinus congestion, musculoskeletal discomfort, two events of cough and three events of acute pulmonary exacerbation. All pulmonary exacerbations were considered serious.
Frequency of occurrence of adverse events occurring in more than one subject in any VX-809 treatment group (listed alphabetically by MedRA term)
Four of 89 (5%) subjects discontinued study drug during the study, one subject in each of the VX-809 dose groups. No placebo-treated subjects withdrew from treatment. All discontinuations were due to the occurrence of respiratory AEs. There were no clinically significant changes in laboratory findings during the study.
PK results
Plasma concentrations of VX-809 were measured by a fully validated bioanalytical method. PK parameters estimated at the four dose levels of VX-809 on days 1 and 28 are summarised in table 3. VX-809 appeared to be slowly absorbed from the gut in subjects with CF with median time to reach maximum concentration (tmax) values ranging from 3 to 4 h across all doses on days 1 and 28. Based on predose PK samples collected from day 1 to day 28, plasma steady-state concentrations appeared to be reached by day 7 at all dose levels with a mean accumulation ratio ranging from 1.7 to 2.0 on day 28 (based on area under the cuve (AUC) over the 24 h dosing period, AUC0–24 h). The time to reach steady state and the extent of accumulation observed in this study were consistent with a terminal half-life approaching 24 h. The estimated mean values of this PK parameter in this study were slightly lower (range from 15 to 18 h) which may be explained by the low number of PK samples collected beyond 24 h on the last dosing day. The estimated oral VX-809 clearance (ie, volume of plasma purified per unit of time) at steady state was relatively low (<2% hepatic blood flow) and comparable for all doses. The estimated volume of distribution of VX-809 at steady state (Vz/F) suggested a potential diffusion of the drug into tissues.
VX-809 Pharmacokinetic parameters following once-daily dosing of VX-809 for 28 days
Maximum (Cmax) and total (AUC0–24 h) exposure to VX-809 increased proportionally with VX-809 dose increases from 25 to 200 mg over the 28 days of treatment. The intersubject variability observed for VX-809 in subjects with CF appeared to be moderate and comparable across all doses for Cmax (30–40%) but with a trend to increase slightly with dose for AUC0–24 h (from 40% to 60%) on day 28.
CFTR bioactivity
The effects of VX-809 on F508del-CFTR processing and function were evaluated throughout the trial. The assessments included measurements of sweat chloride concentration (in all subjects) and NPD (in 71 subjects). The relative amounts of the immature (B-Band) and mature (C-Band) forms of F508del-CFTR, which are markers of protein maturation beyond the endoplasmic reticulum and Golgi, were measured in biopsies of the rectal epithelium in 34 subjects.
Sweat chloride
Figure 1 shows changes in sweat chloride values, which were reduced in a dose-dependent manner (p=0.0013) in VX-809-treated subjects. The reduction in sweat chloride values was rapid and sustained, with measurable changes seen within 7 days of VX-809 dosing (figure 1A). The mean change from baseline in sweat chloride concentration (mmol/l) after 7 days was 2.2 in the placebo group, −0.5 in the 25 mg group, −3.7 in the 50 mg group (95% CI −7.1 to −0.28, p=0.03), −2.3 in the 100 mg group and −6.6 in the 200 mg group (95% CI −10.27 to −2.83, p=0.0008). At day 28, the mean treatment differences from baseline (–placebo) for the 25, 50, 100 and 200 mg groups were +0.10, −4.61, −6.13 (95% CI −12.25 to −0.01) and −8.21 mmol/l (95% CI −14.33 to −2.10), respectively (figure 1B). These differences were statistically significant versus placebo for the 100 mg (p<0.05) and 200 mg (p<0.01) groups. Following 7 days of drug washout, mean sweat chloride values returned to approximately pretreatment levels (figure 1A).

(A) Change in sweat chloride measurements from baseline for placebo- and VX-809-treated subjects. Mean values are shown (± 95% CI, based on analysis of covariance (ANCOVA) analysis) predose, at weekly intervals over the course of treatment, and 1 week following discontinuation of study drug. (B) Sweat chloride change from baseline to day 28 treatment, difference versus placebo (mean (95% CI, based on ANCOVA analysis)). Sweat Cl− changes were seen as early as day 7 of treatment (data not shown), and reached statistical significance for the 100 and 200 mg dose groups (p<0.05 and p<0.01, respectively).
The number of subjects whose sweat chloride values were reduced by the threshold (‘responder’) values was evaluated using predefined and posthoc study criteria. Using a posthoc responder criterion of ≥10 mmol/l reduction, six subjects (38%) in the 200 mg group and six subjects (40%) in the 100 mg group responded compared with none of the placebo-treated patients (p=0.02 for both VX-809 treatment groups). In the 25 and 50 mg dose groups, there were no subjects classified as responders by either criterion. Using the predefined responder criterion, only one subject (6.3%, in the 200 mg dose group) had a ≥20 mmol/l response to VX-809 (p=NS).
Nasal potential difference
Seventy-one subjects underwent NPD measurements throughout the study. There was no significant change in CFTR-dependent NPD parameters (chloride or sodium transport) in any of the dose groups. Supplementary figure 1 summarises the CFTR chloride ion transport parameters (change in chloride-free isoproterenol response) for the VX-809 treatment groups from baseline to day 28 after removing the change in the placebo group (treatment difference).
CFTR B- to C-Band maturation
Thirty-four subjects provided rectal biopsy tissue for evaluation of F508del-CFTR maturation. Among the 33 subjects who were homozygous for the F508del-CFTR allele, no mature C-Band was detected (supplementary figure 2). Only one subject in the highest dose group (VX-809 200 mg) provided rectal biopsy tissue.
Clinical outcomes
Spirometry
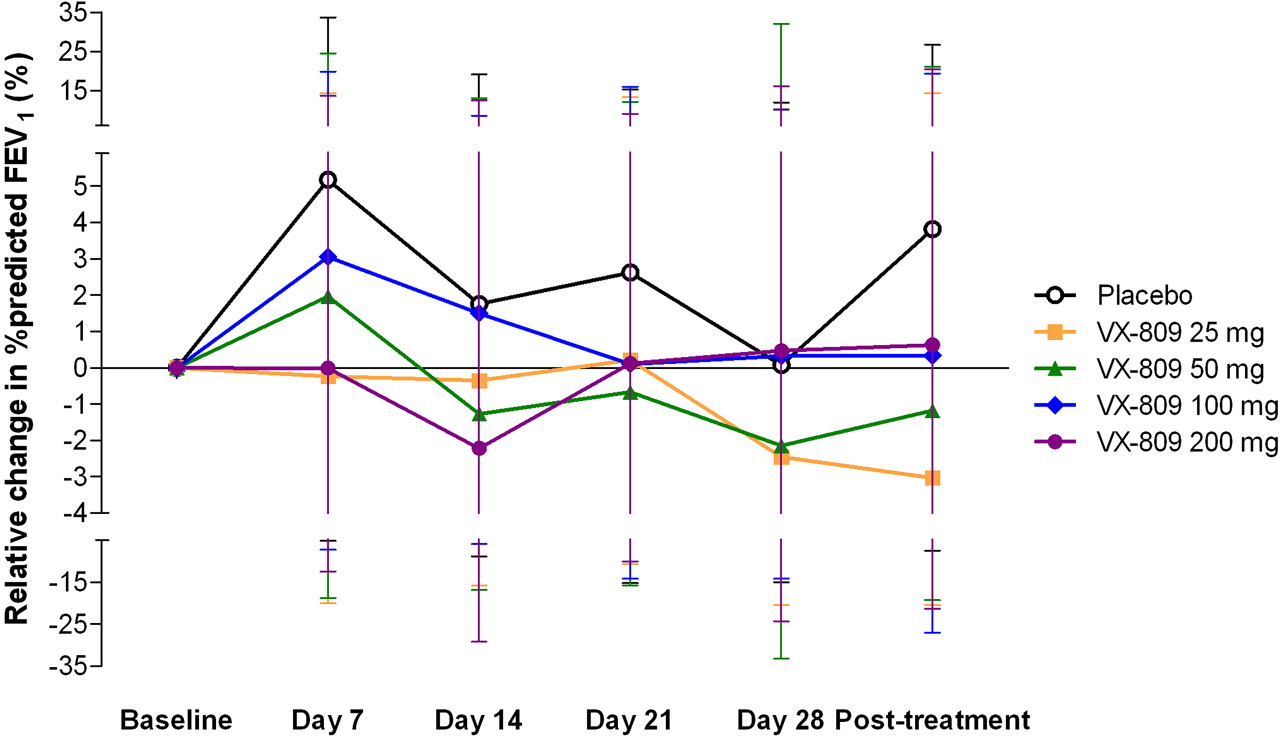
There were no significant changes in lung function (FEV1, FVC, FEF25–75%) in any of the dose groups, including changes in percentage predicted values versus baseline and placebo, or raw measures of litre flow (data not shown). Figure 2 shows the percentage change relative to baseline FEV1 (percentage predicted) for each study group across all study visits. After 28 days of treatment, the mean percentage change from baseline in FEV1 percentage predicted was 0.07, −2.46, −2.15, 0.32 and 0.47 in the placebo, 25, 50, 100 and 200 mg dose groups, respectively (p=NS).
{kind=link}
{kind=link}
Percentage change from baseline in forced expiratory volume in 1 s (FEV1) % predicted (95% CI, based on analysis of covariance (ANCOVA) analysis). No significant changes in FEV1 compared with baseline or placebo were seen for any of the VX-809 dose groups over the course of the study.
Patient-reported outcomes
Supplementary table 1 summarises the change in CFQ-R measures (day 28 vs baseline) across the placebo- and VX-809-treated subjects. There were no clear or sustained changes in the respiratory domain or in any other subdomains of the CFQ-R in any dose group. After 28 days, the respiratory domain score in the placebo group increased by 4.5. After 28 days of treatment in the 25, 50, 100 and 200 mg dose groups, the changes were −5.2, −6.3, −1.30 and +2.2, respectively.
Discussion
These data provide evidence of a safety and tolerability profile sufficient to support further clinical evaluation of VX-809 in subjects with CF homozygous for the F508del-CFTR mutation, the most common CF mutation worldwide. Over a dosing range of 25–200 mg, study participants reported symptoms and AEs that were similar to those in the placebo group, and similar to those commonly found in adult patients with CF. One subject in each of the four dose groups developed a pulmonary exacerbation during the 28 day period of treatment, and the incidence of symptoms such as cough was similar across the dose groups and to that of the placebo group.
Sweat chloride concentration measurements demonstrated modest, statistically significant improvements in VX-809-treated subjects. This effect was dose dependent, sustained during the treatment period and rapidly reversed following discontinuation of VX-809. This suggests that VX-809 increased the chloride transport function of CFTR in the sweat gland, and supports the idea that VX-809 is bioactive in the sweat gland of patients homozygous for the F508del-CFTR mutation. These results also support the hypothesis that small molecule correction of F508del-CFTR is feasible. The results provide support for the use of previously obtained CFTR biomarker data sets for study planning of new CFTR modulators. Using this paradigm, the treatment effects on sweat chloride reported with the CFTR potentiator VX-770 in patients with CF with the G551D-CFTR mutation were successfully used to power the current study to detect improvements in sweat chloride (within-group comparisons and with placebo). It is not clear if the reductions in sweat chloride in this study achieved a peak effect (figure 1), and this raises the question of whether higher doses of VX-809 may produce greater effects on F508del-CFTR. The peak corrective effect of VX-809 on F508del-CFTR in primary HBE cells has been estimated at 3 μM (F. Van Goor, personal communication, 2011). Such concentrations of VX-809 were observed in the plasma of patients in the 100 and 200 mg dose cohorts at steady state. However, the drug exposure levels achieved in patient target tissues at these doses of VX-809 is not known.
The other biomarkers of CFTR activity (NPD, immunoblot from rectal biopsy tissue) failed to demonstrate changes over the course of VX-809 treatment. This may reflect the trial's lack of power to detect a predicted change in these measurements based on in vitro results with VX-809 in F508del/F508del primary HBE cells (maximum of ∼15% of wild-type CFTR function).17 The intrinsically greater variability of NPD measurement (potentially compounded by the large number of NPD sites used in this trial (n=19)) led to a greater likelihood of Type 2 error in analysing biomarker responses. To minimise a Type 2 error and attain an 80% power to detect a chloride-free isoproterenol response approximating 30% of that seen with VX-770 in patients with CF with the G551D-CFTR mutation, the current study would have required >50 subjects per study group undergoing the NPD.17 22
In vitro studies also suggest that measurement of the effect of VX-809 on CFTR may be below the lower limits of sensitivity of immunoblot assays performed on small biopsy samples.17 Pretrial studies indicated that the assay developed here was capable of detecting ∼10% of C-Band CFTR relative to wild-type levels (ex vivo dilution experiments using human rectal biopsy specimens obtained from non-CF study subjects; see online supplement, figure 2B). The sensitivity of this assay to detect F508del-CFTR correction relative to functional measurements in vivo is unknown, but in vitro studies in a variety of model systems suggest that biochemical detection of F508del-CFTR correction may be less robust than functional measures in intact epithelia.17 23 The failure to detect VX-809 effects on F508del-CFTR outside of the sweat gland could also reflect tissue-specific differences in VX-809 bioavailability and/or F508del-CFTR responsiveness to VX-809 treatment.
No improvements in clinical outcomes, including lung function and quality of life (CFQ-R), were observed over the course of this study. The trial was not powered to detect such improvements in these measures, and longer or larger trials of VX-809 (alone or in combination with CFTR potentiators) may be necessary to determine the clinical effect of these CFTR modulators.
Previous trials of systemically dosed CFTR modulators have frequently described discordant effects on CFTR-dependent biomarkers and pulmonary outcome measures. For example, PTC124 has been shown to have detectable bioactivity by NPD over 2 weeks of treatment in patients with CF possessing premature termination codons in CFTR, while sweat chloride measurements remained unchanged.24 Improvements in lung function and cough frequency were not observed until months of treatment were completed.25 Systemic gentamicin has also been shown to suppress PTCs26 and improve NPD parameters in two pilot studies, but effects on sweat chloride were predominantly limited to a subset of patients with the Y122X mutation.27 28 Using a separate CFTR modulator strategy, Rubenstein and colleagues treated F508del-CFTR homozygous patients with CF with the F508del-CFTR modulator 4-phenyl butyrate. Improvements in NPD-dependent chloride secretion were seen, but there were no effects on sweat chloride.29 The reasons for discrepancies in CFTR biomarkers across different CFTR modulator strategies are not clear, but suggest that organ effects may vary due to tissue drug availability, CFTR regulation in different cell types or the responsiveness of mutant CFTR across different tissue compartments and modulator strategies. These uncertainties should be considered in future study planning, as should the selection and continued development of CFTR biomarkers to demonstrate study drug bioactivity.
In conclusion, the results provide support for the continued evaluation of VX-809 in patients with CF with the F508del-CFTR mutation. This study demonstrated that VX-809 has an acceptable safety profile. It also provided evidence of the effect of VX-809 on improving CFTR function based on dose-dependent reductions in F508del-CFTR activity in the sweat gland using a CFTR corrector administered orally to subjects with CF, and further investigation is warranted to evaluate the potential of this therapeutic strategy to increase F508del-CFTR activity.
Acknowledgments
The authors would like to acknowledge the research coordinators who participated in this trial (Ginger Reeves, University of Alabama at Birmingham; Jill Hammond, University of Colorado Denver; Carolyn Chapman, Erin Felling and Karen Callahan, Johns Hopkins Medical Institutions; Alan Genatossio, University of Washington; Bobbi Ksenich, Rainbow Babies and Children's Hospital; Julie Avolio and Katherine Keenan, Hospital for Sick Children, Toronto; Myra Slutsky at St Michaels's Hospital Toronto; Terri Johnson, Nationwide Children's Hospital; Cathy Powers, Northwestern University; Colleen Dunn, Stanford University; Patricia Burks, Washington University School of Medicine; Elizabeth Hartigan RN, MPH; Adrienne DeRicco, and Sandy Hurban, UPMC); the site NPD operators (Gina Sabbatini, University of Alabama at Birmingham; Lisa Monchill, University of Colorado Denver; Christina Kubrak, Children's Hospital of Philadelphia; Douglas Walker, Johns Hopkins Medical Institutions; Zoe Davies, Stanford University; Louis Boitano, University of Washington; Teresinha Leal, Université de Louvain; Denise Rodgers, Washington University School of Medicine); Linda Gabel and the Chemistry staff at Colorado Children's Hospital for performing sweat chloride analyses; members of the Center for CFTR Detection which provided NPD interpretation and oversight (Heather Hathorne, MAE, RRT, and Bo Liu, MPH); The authors would like to acknowledge the following co-primary investigators of the study: Dr Felix Ratjen at the Hospital for Sick Children, Toronto; Dr Elizabeth Tullis at St Michael's Hospital, Toronto. The authors would like to acknowledge the following employees of Vertex Pharmaceuticals: Hong Lu, Paul Panorchan and Slawa Rockiki for their pharmacokinetic support, Yanyan Cui for drug metabolism assistance, Lynn Anderson for statistical programming assistance, Terry Appleton for clinical data management, Yong Gu and Catherine Phillips for assistance with biomarker analyses, Marjorie Layman (Vertex Pharmaceuticals) for study document writing support, Eric Olson for strategic guidance of the VX-809 programme, Christopher Simard for patient safety analysis, Jaclyn Verrow and Mark Bruce for clinical operations support, and Adrienne Aiello for coordinating the editing and submission of the manuscript.
References
Supplementary materials
Web Only Data thoraxjnl-2011-200393
Files in this Data Supplement:
Footnotes
See Editorial, p 4
Funding Supported by Vertex Pharmaceuticals and grants CTSA UL1 RR024134 to the Children's Hospital of Philadelphia; NIH 1UL1 RR025744 to Stanford University; NIH K23 DK075788 to SMR; IUL1 RR025777 to Dr Guay-Woodford; CFF CLANCY09YO to SMR; AMIN09YO to RSA; NIH/NCRR/CTSA Grant UL1 RR024153 to the University of Pittsburgh; UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research; NIH grants UL1 RR024989 and P30 DK27651 to Case Western Reserve University; UL1 RR025747 from the NCRR. M01 RR00400 from the NCRR from Harvard Catalyst/The Harvard Clinical and Translational Science Center (NIH Award #UL1 RR 025758 and financial contributions from Harvard University and its affiliated academic health care centres). UL1 RR 025005 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research to the Johns Hopkins School of Medicine. CTSA (Clinical and Translational Science Award) Grant number is 1UL1 RR025014 to the University of Washington. CFFT Durie 06A0. CTSA grant: UL1RR025747 to the University of North Carolina Chapel Hill. CTSA 1UL1 RR025780 to University of Colorado. Washington University ICTS/CTSA Grant Number UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research to Washington University in St. Louis. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centres, the NCRR or the NIH.
Correction notice This article has been corrected since it was published Online First. The author name Kris DeBoeck has been corrected to Kris De Boeck.
Competing interests None.
Ethics approval This study was conducted with the approval of the Institutional Review Board at each site.
Provenance and peer review Not commissioned; externally peer reviewed.