Article Text

Abstract
Rationale Elevated plasma and bronchoalveolar lavage fluid plasminogen activator inhibitor 1 (PAI-1) levels are associated with adverse clinical outcome in patients with pneumonia caused by Pseudomonas aeruginosa. However, whether PAI-1 plays a pathogenic role in the breakdown of the alveolar–capillary barrier caused by P aeruginosa is unknown.
Objectives The role of PAI-1 in pulmonary host defence and survival during P aeruginosa pneumonia in mice was tested. The in vitro mechanisms by which P aeruginosa causes PAI-1 gene and protein expression in lung endothelial and epithelial cells were also examined.
Methods and results PAI-1 null and wild-type mice that were pretreated with the PAI-1 inhibitor Tiplaxtinin had a significantly lower increase in lung vascular permeability than wild-type littermates after the airspace instillation of 1×107 colony-forming units (CFU) of P aeruginosa bacteria. Furthermore, P aeruginosa in vitro induced the expression of the PAI-1 gene and protein in a TLR4/p38/RhoA/NF-κB (Toll-like receptor 4/p38/RhoA/nuclear factor-κB) manner in lung endothelial and alveolar epithelial cells. However, in vivo disruption of PAI-1 signalling was associated with higher mortality at 24 h (p<0.03) and higher bacterial burden in the lungs secondary to decreased neutrophil migration into the distal airspace in response to P aeruginosa.
Conclusions The results indicate that PAI-1 is a critical mediator that controls the development of the early lung inflammation that is required for the activation of the later innate immune response necessary for the eradication of P aeruginosa from the distal airspaces of the lung.
- PAI-1
- P aeruginosa
- pneumonia
- RhoA
- acute lung injury
- airway epithelium
- bacterial infection
Statistics from Altmetric.com
Key messages
What is the key question?
The role of PAI-1 in pulmonary host defence and survival during P aeruginosa pneumonia in mice is not known.
What is the bottom line?
PAI-1 is a critical mediator that controls the development of the early lung inflammation that is required for the activation of the later innate immune response necessary for the eradication of P aeruginosa from the distal airspaces of the lung.
Why read on?
Although associated with an early attenuation of the severity of pulmonary oedema formation, in vivo disruption of PAI-1 signalling is associated with higher mortality at 24 h secondary to decreased neutrophil migration into the distal airspace in response to P aeruginosa.
Introduction
Pseudomonas aeruginosa is an opportunistic pathogen that causes lethal pneumonia in immunocompromised individuals and in critically ill patients.1 2 P aeruginosa pneumonia is characterised by a shift in the balance between procoagulant and anticoagulant/fibrinolytic activity in the alveoli followed by intra-alveolar fibrin deposition. Early procoagulant activity in the alveolar space may exert beneficial effects by decreasing the protein leakage through the alveolar–capillary barrier caused by P aeruginosa. However, excessive fibrin deposition causes deleterious effects by increasing the lung inflammatory response and vascular permeability. When fibrin is present in the alveolar space, it is usually degraded by plasmin, a proteolytic enzyme that is present in the lung tissue as an inactive zymogen. However, decreased fibrinolytic activity has been reported in the airspaces of the lungs of patients with P aeruginosa pneumonia.3
The plasminogen activator inhibitor 1 (PAI-1), a member of the serine protease inhibitors superfamily,4 which is expressed by endothelial cells, epithelial cells, monocytes and macrophages, is the principal inhibitor of tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) and thus inhibits fibrinolysis. PAI-1 levels have been shown to be elevated in the airspace and plasma of patients with ventilator-associated pneumonia (VAP), aspiration pneumonia and acute lung injury (ALI),5–7 and to correlate with severe disease and adverse clinical outcome.8 Furthermore, bronchoalveolar lavage (BAL) fluid levels of PAI-1 are higher in patients with P aeruginosa-induced VAP than those found in other infections9 and increased BAL levels of PAI-1 were associated with increased mortality in a cohort of intensive care patients with P aeruginosa VAP,10 suggesting a role for PAI-1 in the development of severe P aeruginosa VAP. However, these clinical associations do not establish a direct causal link between PAI-1 and the development of more severe P aeruginosa pneumonia, particularly because contrasting results have been reported with PAI-1 gene deletion in several experimental models of bacterial pneumonia in mice.11 12 PAI-1 gene deletion has also been shown to be protective in non-infectious experimental models of lung injury induced by ischaemia–reperfusion or hyperoxia.13 14 Thus, it is not clear whether PAI-1 itself is beneficial or deleterious for the host during P aeruginosa pneumonia, and answering this question constitutes the goal of the present study.
Material and methods
More details are available in the online supplement.
Reagents
All cell culture media were prepared by the University of California San Francisco Cell Culture Facility using deionised water and analytical grade reagents.
Cell culture
Bovine pulmonary arterial endothelial cells (BPAECs; ATCC, CCL-209; passages all <8) were cultured, as previously described.15
Human lung microvascular endothelial cells (HMVECs; Lonza; CC-064) were cultured either on transwells (Costar 3495, 2×105 cells per filter) for 4 days or in 6-well plates until they formed confluent monolayers.
Primary rat alveolar epithelial type II (ATII) cells were isolated and cultured as previously described.16
Preparation of P aeruginosa
The wild-type, PAK strain of P aeruginosa was a kind gift from Dr Stephen Lory at Harvard University, Massachusetts. The PA103 strain of P aeruginosa was generously provided by Dr Dara Frank at the Medical College of Wisconsin. The preparation of P aeruginosa was performed as previously described.15
Measurement of transendothelial albumin flux
Transendothelial albumin flux across BPAECs and HMVECs was measured as previously described.15
Western blot analysis
Western blot analysis was performed as described previously.17
Primers and probes
Real-time reverse transcription–PCR primers and probes were designed using Primer Express software (PE-Applied Biosystems, Warrington, UK) and purchased from Biosearch Technologies Novato, CA, USA.
Quantitative real-time RT–PCR
Quantitative real-time RT–PCR analyses were performed as described previously.17
RhoA and Rac-1 activity assay in cells
Rac-1 and RhoA activities were determined using a luminescence-based G-LISA Rac-1 and RhoA activation assay (Cytoskeleton, Denver, Colorado, USA).
Cell treatment with exogenous PAI-1
This was performed as described in our previous study.18
Cell viability assay
Cell viability was measured by the Alamar Blue assay after exposure to the various experimental conditions.
Mice
Wild-type C57BL/6 (C57BL/6J #000664) and PAI-1 null (strain: C57BL/6J #000257) mice were purchased from Jackson Laboratories Bar Harbor, ME, USA.
Pneumonia model
This model was produced and used as described in our previous study.19
Lung vascular permeability measurement
Lung excess lung water (ELW, μl) and extravascular plasma equivalent (EVPE, μl) were measured as previously described.20
BAL, cell count and keratinocyte-derived chemokine (KC) measurements
BAL fluid was collected by infusing 1 ml of sterile phosphate-buffered saline (containing 5 mM EDTA) into the lungs of the mice after tracheal cannulation, as previously described.21 Cytospin and KC measurements were performed after centrifugation.
Bacterial cultures from the lung homogenates
The lungs were homogenised in sterile containers using a tissue homogeniser (Tissue Tearor model 985-370, Biospec products, Racine, Wisconsin, USA). The homogenates were serially diluted and plated in triplicate on sheep-blood agar plates.
Lung myeloperoxidase measurement
After homogenisation, the lungs were clarified by centrifugation. The measurement of myeloperoxidase was performed according to the manufacturer's protocol.
Survival protocol
Twenty wild-type mice were randomly assigned to two groups (untreated mice n=10; PAI-039-treated mice n=10). A third group was composed of 10 PAI-1 null mice. Mice were exposed to P aeruginosa as described previously.19
Statistical analysis
All data are summarised as mean ± SD. For the statistical analysis we used Statview Version 5.0 and MedCalc Version 7.2.0.2. The normal distribution was verified using the Kolmogorov–Smirnov test. For normally distributed series, comparison of three or more experimental groups was performed by one-way analysis of variance (ANOVA). For non-normally distributed series or series of three measurements, the Mann–Whitney test was used to compare two experimental conditions. A Kaplan–Meier analysis followed by a logrank (Mantel–Cox) test was used to compare the survival between the three experimental groups of mice at 48 h. A p value of <0.05 was considered statistically significant.
Results
PAI-1 is an essential component of the pulmonary host response during the early phase of P aeruginosa pneumonia in mice
We first found that there was a time-dependent increase in plasma and BAL fluid levels of PAI-1 in C57Bl6 mice instilled with P aeruginosa (figure 1). We also found that the increase in lung vascular permeability caused by the airspace instillation of two different strains of P aeruginosa (PA103 that produces exoU and PAK that does not produce exoU) was significantly attenuated in PAI-1 null mice (figure 2A–D). These results were confirmed by treating the mice with Tiplaxtinin (also called PAI-039), a newly developed pharmacological inhibitor of PAI-1 (figure 2E,F). We next showed that PAI-1 inhibition using PAI-039 (11 μM) significantly decreased PAK-induced paracellular permeability in both human and bovine pulmonary endothelial cells (figure 3A,B). Figure 3C,D also shows that PAI-1 inhibition significantly attenuated PAK-induced RhoA activation in both BPAEC and HMVEC monolayers. Finally, the Alamar Blue assay showed that exposure to PAK for 3 h did not decrease the viability of lung endothelial or alveolar epithelial cells (data not shown).
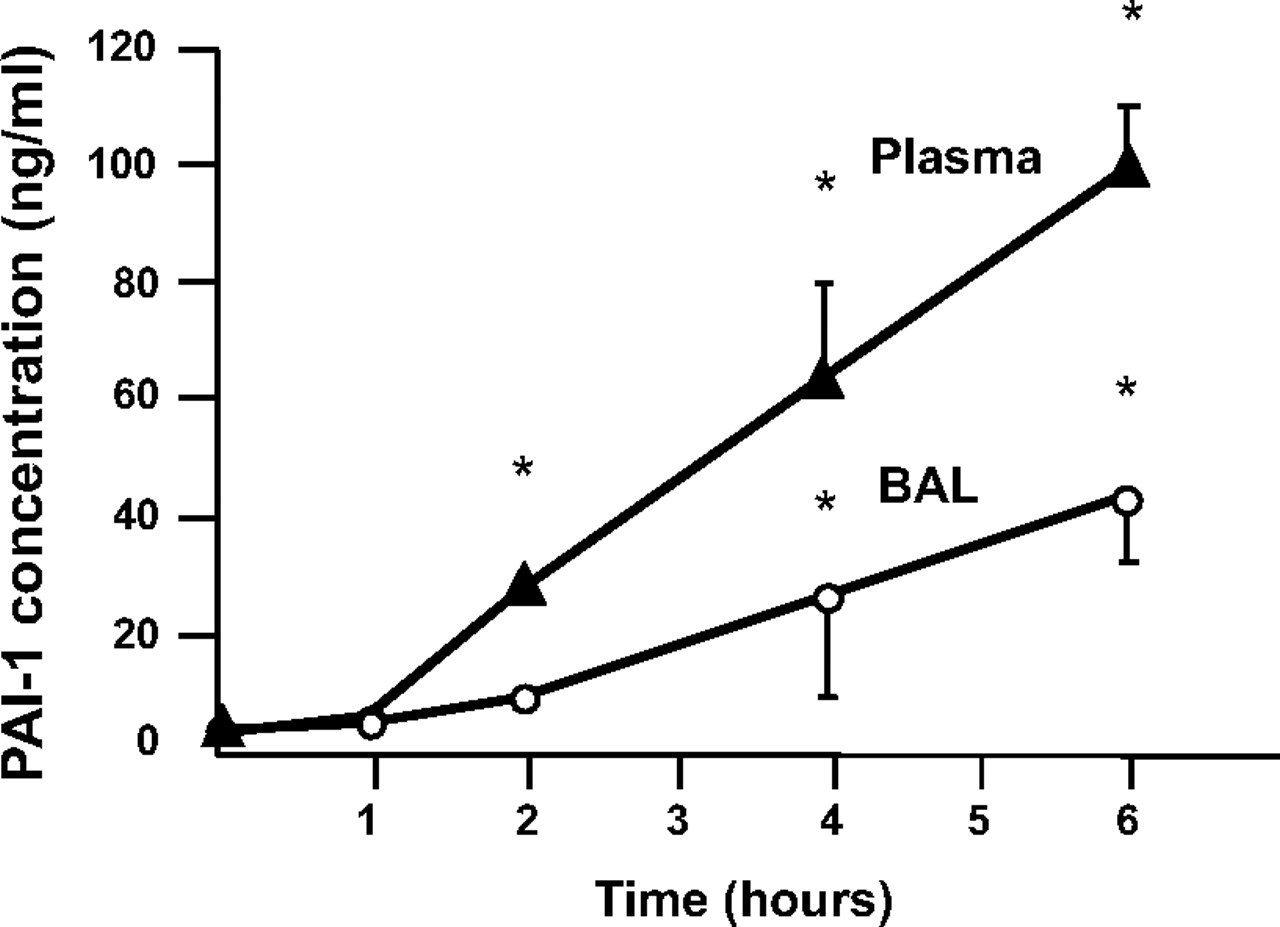
Airspace instillation of P aeruginosa causes a time-dependent increase in plasma and bronchoalveolar lavage (BAL) fluid levels of plasminogen activator inhibitor-1 (PAI-1) in mice. C57BL/6 mice were instilled with P aeruginosa (PAK strain, 1×107 colony-forming units) or its vehicle. Data are mean ± SD (n=8 mice in each experimental group); *p<0.05 from time 0 (control mice).

Plasminogen activator inhibitor-1 (PAI-1) gene deletion or pharmacological inhibition attenuates the increase in lung vascular protein permeability caused by the airspace instillation of P aeruginosa in mice. (A–D) Wild-type or PAI-1 (–/–) (C57BL/6 strain) mice were instilled with P. aeruginosa (PAK strain, 1×107 colony-forming units (CFU)) or its vehicle. Four hours later, mice were euthanised and excess lung water (ELW) and lung vascular permeability (extravascular plasma equivalent (EVPE)) to protein were measured as described in the Materials and methods section. For all experiments, results are shown as mean ± SD (n=8 mice in each experimental group); *p ≤0.05 from controls; #p ≤0.05 from wild-type mice instilled with P aeruginosa. (E–F) C57BL/6 mice were instilled with P aeruginosa (PAK strain, 1×107 CFU or PA103 strain 5×105 CFU) or their vehicle. Mice were treated with a specific PAI-1 inhibitor Tiplaxtinin (PAI-039) (30 mg/kg) or its vehicle given intraperitoneally twice, 1 h before and at the time of airspace instillation of P aeruginosa. For all experiments, results are shown as mean ± SD (n=8 mice in each experimental group); *p ≤0.05 from controls; #p ≤0.05 from mice instilled with P aeruginosa and treated with Tiplaxtinin vehicle.

Specific plasminogen activator inhibitor-1 (PAI-1) inhibition attenuates P aeruginosa-induced increase in protein permeability and RhoA activity in bovine pulmonary artery and human microvascular lung endothelial cell monolayers. (A–D) Bovine pulmonary artery (BPAEC) and human microvascular (HMVEC) lung endothelial cell monolayers were treated with the wild-type P aeruginosa strain PAK (bacterial to bovine cell ratio: 1:2) or its vehicle for 30 min (RhoA activity) or 4 h (permeability measurement). Cell monolayers were pretreated with Tiplaxtinin (PAI-039) (11 μM) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). All experiments were performed at least in triplicate and repeated three times. Results are shown as mean ± SD; *p≤0.05 from controls; #p≤0.05 from cell monolayers treated with PAK and PAI-039 vehicle.
P aeruginosa (PAK) induces PAI-1 expression in pulmonary endothelial and alveolar epithelial cells via a RhoA-dependent mechanism
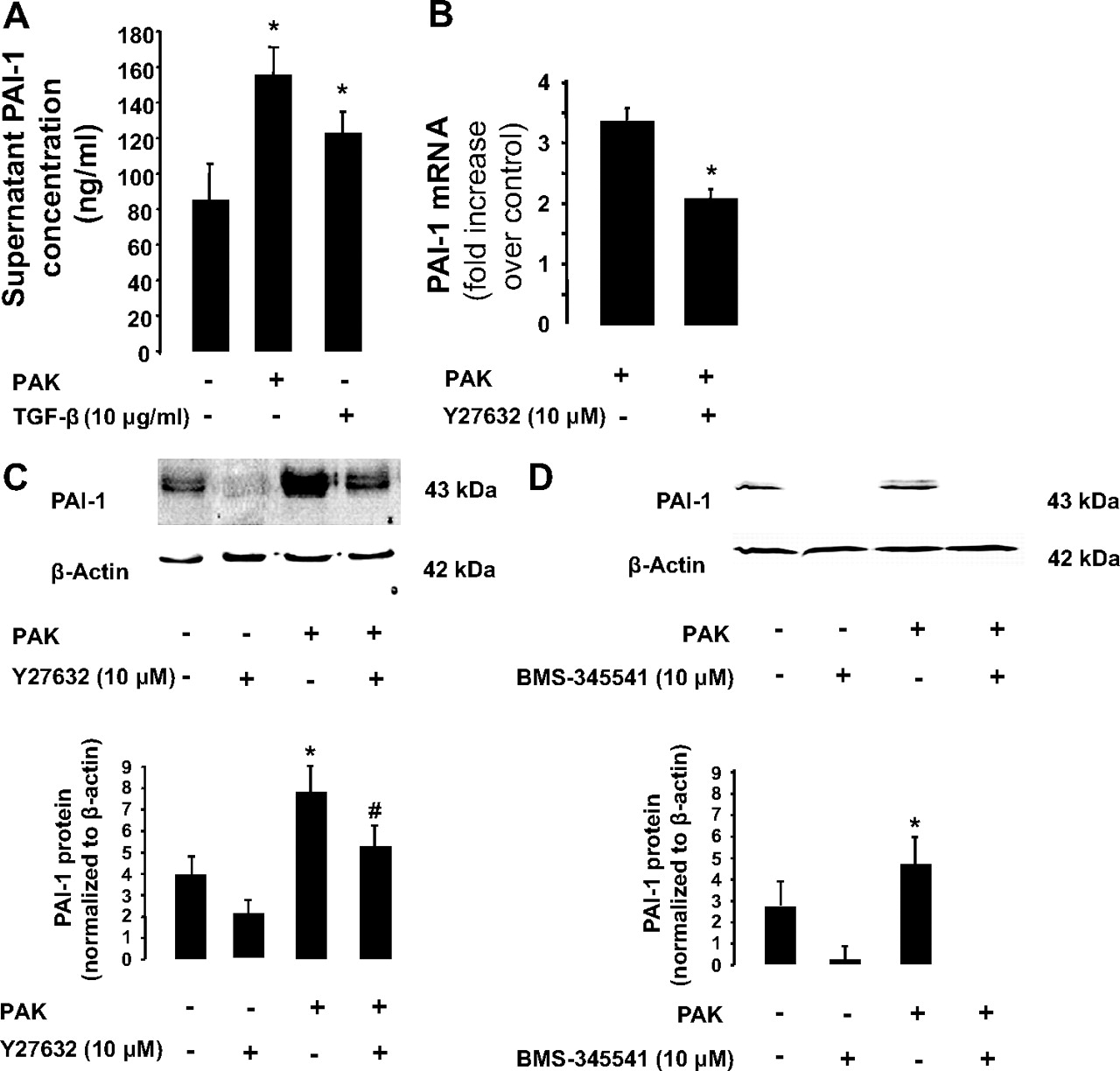
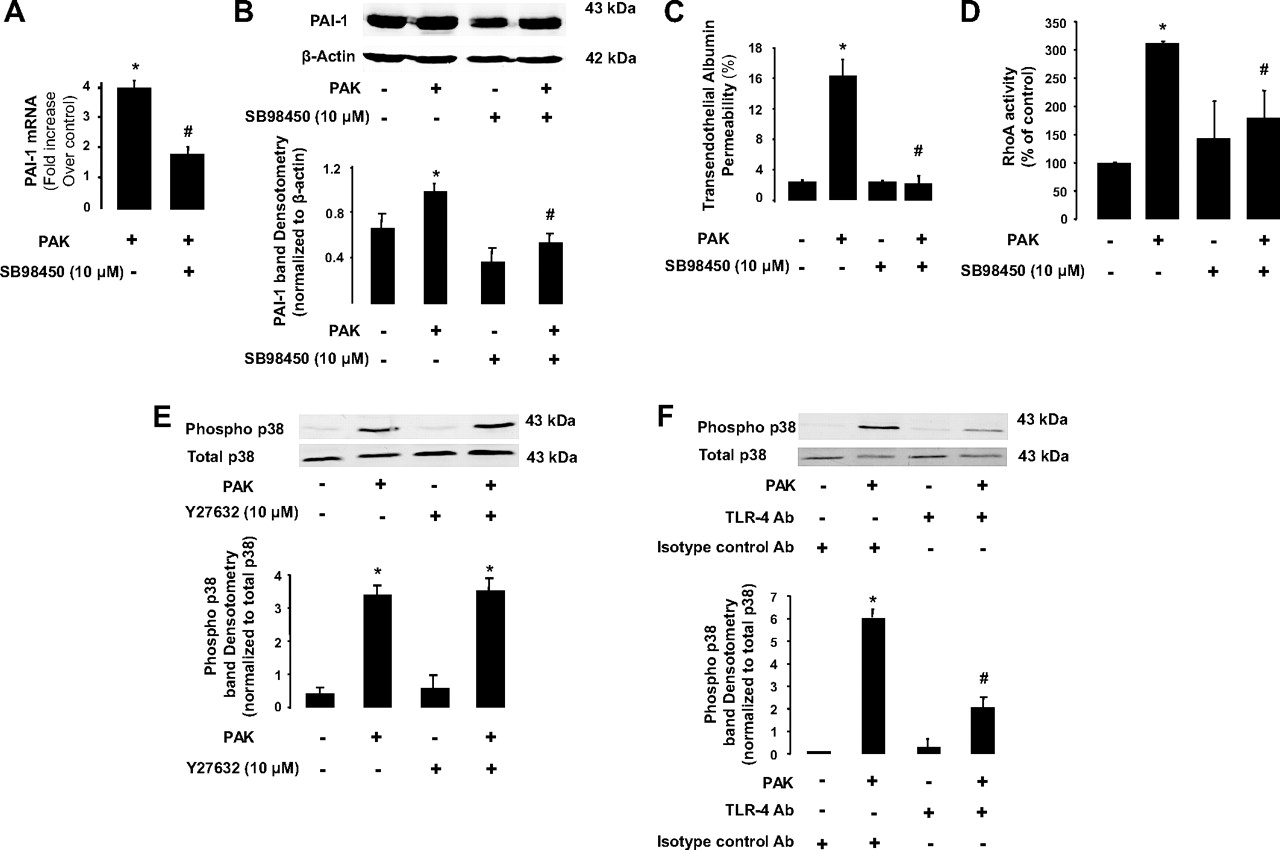
Exposure of endothelial cells to PAK or transforming growth factor β1 (TGFβ1) resulted in the release of PAI-1 into the cell supernatant (figure 4A). Exposure to PAK also resulted in an increase in PAI-1 gene and protein expression (figure 4B,C). Furthermore, pretreatment of BPAEC monolayers with a ROCK inhibitor (Y-27632, 10 μM) or with a nuclear factor-κB (NF-κB)-dependent kinase (IκB) inhibitor (BMS-345541, 10 μM) significantly decreased PAI-1 protein expression in BPAEC monolayers (figure 4C,D). We also found that inhibition of p38 mitogen-activated protein (MAP) kinase activity with SB98450 (10 μg/ml) significantly decreased PAK-mediated induction of PAI-1 gene and protein expression (figure 5A,B) and protein permeability across BPAEC monolayers induced by P aeruginosa (figure 5C). Interestingly, inhibition of the p38 MAP kinase decreased PAK-mediated RhoA activation whereas the inhibition of the RhoA-dependent kinase by its inhibitor Y-27632 did not prevent P aeruginosa-induced p38 MAP kinase activation (figure 5D,E). Next, we found that PAK-induced p38 MAP kinase activation was largely dependent on activation of Toll-like receptor 4 (TLR4) by this bacterium (figure 5F). Finally, we found that exposure of BPAEC monolayers to human recombinant PAI-1 caused a reversal of the RhoA/Rac-1 activity ratio that resulted in the activation of RhoA and a corresponding inhibition of Rac-1 which could further increase the direct effect of P aeruginosa on lung vascular permeability (figure 6A,B).

P aeruginosa causes plasminogen activator inhibitor-1 (PAI-1) gene and protein expression via a RhoA- and nuclear factor-κB (NF-κB)-dependent mechanism in bovine pulmonary artery epithelial cell (BPAEC) monolayers. (A) P aeruginosa causes the extracellular release of PAI-1 protein by BPAEC monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 24 h (bacterial to bovine cell ratio: 1:5). Transforming growth factor β1 (TGFβ1) (10 ng/ml) served as a positive control. (B and C) P aeruginosa causes PAI-1 gene and protein expression via a RhoA-dependent mechanism in BPAEC monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 4 h (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with the ROCK inhibitor Y27632 (10 μM) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). (D) P aeruginosa causes PAI-1 protein expression via an NF-κB-dependent mechanism in BPAEC monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 4 h (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with the NF-κB-dependent kinase (IκB) inhibitor BMS-345541 (10 μM) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). All experiments were performed at least in triplicate and repeated at least three times. For western blots, a representative blot is shown. Three additional experiments gave comparable results. Results are shown as mean ± SD; *p≤0.05 from negative or positive controls; #p≤0.05 from cell monolayers treated with PAK and Y27632 or BMS-345541 vehicle.
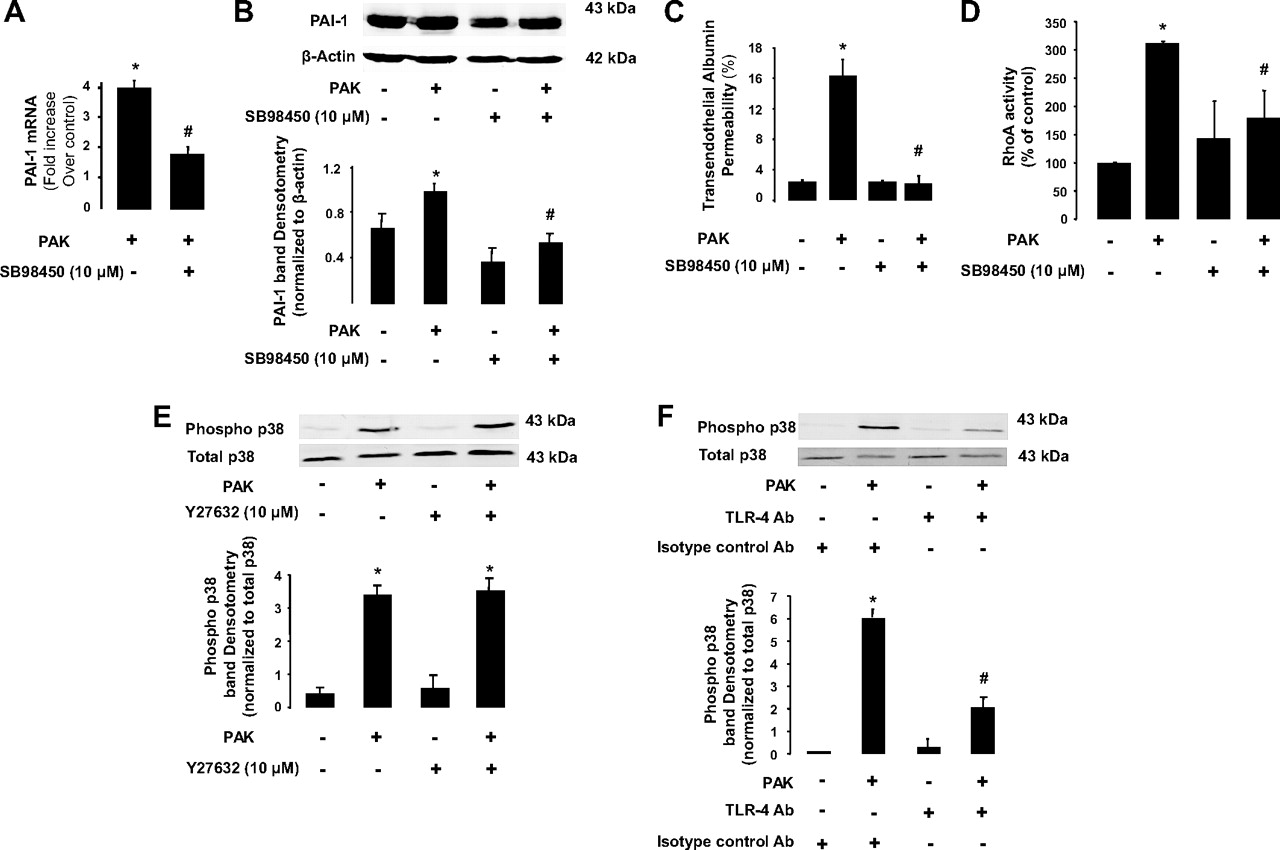
P aeruginosa causes plasminogen activator inhibitor-1 (PAI-1) gene and protein expression via a p38 mitogen-activated protein (MAP) kinase- and Toll-like receptor 4 (TLR4)-dependent mechanism in lung endothelial cell monolayers. (A and B) P aeruginosa causes PAI-1 gene and protein expression via a p38 MAP kinase-dependent mechanism in bovine pulmonary artery epithelial cell (BPAEC) monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 4 h (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with the p38 MAP kinase inhibitor SB88450 (10 μg/ml) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). (C) Specific p38 MAP kinase inhibition attenuates P aeruginosa-induced increase in protein permeability in BPAEC monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 4 h (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with the p38 MAP kinase inhibitor SB88450 (10 μg/ml) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). (D) Pretreatment with the p38 MAP kinase inhibitor SB88450 attenuates the P aeruginosa-mediated RhoA activation in BPAEC monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 30 min (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with the p38 MAP kinase inhibitor SB88450 (10 μg/ml) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). (E) Pretreatment with the ROCK inhibitor Y27632 does not attenuate the P aeruginosa-mediated p38 MAP kinase phosphorylation in BPAEC monolayers. BPAEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 4 h (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with the ROCK inhibitor Y27632 (10 μM) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). (F) Pretreatment with a specific blocking antibody (Ab) against TLR4 attenuates the P aeruginosa-mediated p38 MAP kinase phosphorylation in human lung microvascular endothelial cell (HMVEC) monolayers. HMVEC monolayers were treated with the wild-type P aeruginosa strain PAK or its vehicle for 4 h (bacterial to bovine cell ratio: 1:2). Cell monolayers were treated with a specific blocking Ab against TLR4 or its isotype control Ab for 1 h prior to stimulation with P aeruginosa (PAK). All experiments were performed at least in triplicate and repeated at least three times. For western blots, a representative blot is shown. Three additional experiments gave comparable results. Results are shown as mean ± SD; *p≤0.05 from negative or positive controls; #p≤0.05 from cell monolayers treated with PAK and SB98450 vehicle or a control isotype Ab.

Recombinant plasminogen activator inhibitor-1 (PAI-1) increases RhoA and inhibits Rac-1 activity in bovine pulmonary artery epithelial cell (BPAEC) monolayers. (A and B) BPAEC monolayers were treated with human recombinant PAI-1 (20 μg/ml) or its vehicle for 30 min. All experiments were performed at least in triplicate and repeated at least three times. Results are shown as mean ± SD; *p≤0.05 from controls.
As figure 1 shows, there was a time-dependent increase in PAI-1 BAL fluid levels after airspace instillation of PAK in mice. Therefore, we also examined whether there was an increase in PAI-1 protein expression in alveolar epithelial cell monolayers exposed to P aeruginosa. Figure 7 shows that PAI-1 protein was significantly increased after PAK exposure (4 h) and that PAK-mediated induction of PAI-1 expression was RhoA dependent (figure 7).

P aeruginosa causes plasminogen activator inhibitor-1 (PAI-1) protein expression via a RhoA-dependent mechanism in primary cultures of alveolar epithelial type II cell monolayers. Rat alveolar epithelial type II cell monolayers cultured on transwells with an air–liquid interface were treated with the wild-type P aeruginosa strain PAK or its vehicle for 3 h (bacterial to rat cell ratio: 1:2). Cell monolayers were treated with the ROCK inhibitor Y27632 (10 μM) or its vehicle for 1 h prior to stimulation with P aeruginosa (PAK). A representative western blot is shown. Three additional experiments gave comparable results. Results are shown as mean ± SD; *p≤0.05 from controls; #p≤0.05 from cell monolayers treated with PAK and Y27632 vehicle.
PAI-1 deletion and inhibition inhibits the lung innate immune response and increases mortality during P aeruginosa pneumonia in mice
We next examined the effect of PAI-1 deletion or inhibition on bacterial clearance, mortality and lung innate immune response using our murine model of P aeruginosa pneumonia. PAI-1 deletion or inhibition was associated with a significant decrease in lung bacterial clearance (figure 8A,B) and higher mortality (figure 8C). In the next series of experiments, we examined whether the decreased bacterial clearance observed in mice with disrupted PAI-1 signalling was caused by a defect in neutrophil migration into the lung in response to the airspace instillation of P aeruginosa. We found that PAI-1 null mice or wild-type mice treated with PAI-039 had a decreased neutrophil recruitment into the lung in response to the airspace instillation of P aeruginosa (figure 9A,B). Disruption of PAI-1 signalling was also associated with a lower myeloperoxidase activity in the lungs (figure 9C) and lower BAL fluid levels of KC, a major murine neutrophil chemokine (figure 9D).

Plasminogen activator inhibitor-1 (PAI-1) gene deletion or pharmacological inhibition decreases lung bacterial clearance and increases mortality in a mouse model of P aeruginosa pneumonia. (A) Wild-type or PAI-1 (–/–) (C57BL/6 strain) mice were instilled with P aeruginosa (PAK strain, 1×107 colony-forming units (CFU)) or its vehicle. Wild-type mice were treated with a specific PAI-1 inhibitor Tiplaxtinin (PAI-039) (30 mg/kg) or its vehicle given intraperitoneally twice, 1 h before and at the time of airspace instillation of P aeruginosa. Mice were studied for 8 h. For all experiments, results are shown as mean ± SD (n=6 mice in each experimental group); *p≤0.05 from wild-type mice treated with Tiplaxtinin vehicle. (B) Low-power photomicrographs of the cytospin of bronchoalveolar lavage fluids from wild-type or PAI-1 (–/–) (C57BL/6 strain) mice stained with H&E. Wild-type mice were treated with a specific PAI-1 inhibitor Tiplaxtinin (PAI-039) (30 mg/kg) or its vehicle given intraperitoneally twice, 1 h before and at the time of airspace instillation of P aeruginosa. Mice were studied for 8 h. One representative cytospin for each condition is shown. Three additional experiments gave comparable results. (C) Wild-type or PAI-1 (–/–) (C57BL/6 strain) mice were instilled with P aeruginosa (PAK strain, 1×107 CFU) or its vehicle. Wild-type mice were treated with a specific PAI-1 inhibitor Tiplaxtinin (PAI-039) (30 mg/kg) or its vehicle given intraperitoneally twice, 1 h before and at the time of airspace instillation of P aeruginosa. Kaplan–Meier survival analysis was performed (n=10 mice in each experimental group). Mice with a gene deletion or pharmacological inhibition of PAI-1 died significantly earlier than the wild-type mice after airspace instillation of P aeruginosa and treatment with Tiplaxtinin vehicle.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Plasminogen activator inhibitor-1 (PAI-1) gene deletion or pharmacological inhibition attenuates neutrophil recruitment into the airspace and bronchoalveolar lavage (BAL) fluid levels of keratinocyte-derived chemokine (KC) in a mouse model of P aeruginosa pneumonia. (A–D) Wild-type or PAI-1 (–/–) (C57BL/6 strain) mice were instilled with P aeruginosa (PAK strain, 1×107 colony-forming units (CFU)) or its vehicle. Wild-type mice were treated with a specific PAI-1 inhibitor Tiplaxtinin (PAI-039) (30 mg/kg) or its vehicle given intraperitoneally twice, 1 h before and at the time of airspace instillation of P aeruginosa. Mice were studied for 8 h. BAL was performed, and cells were counted. and KC levels measured as described in the Materials and methods sections. For all experiments, results are shown as mean ± SD (n=6 mice in each experimental group); *p≤0.05 from wild-type mice treated with Tiplaxtinin vehicle. MPO, myeloperoxidase.
Discussion
A growing body of literature has highlighted clinical correlations between elevated plasma, BAL or pulmonary oedema fluid levels of PAI-1 and severe disease, as well as adverse clinical outcome in both patients with ALI and with pneumonia caused by P aeruginosa.8–10 22 Decreased airspace fibrinolysis in patients with pneumonia has mainly been attributed to an elevation in PAI-1 activity11 and precedes the clinical recognition of VAP by several days in critically ill patients.5 However, it was still unknown whether there is a role for PAI-1 in lung injury induced by bacteria, such as P aeruginosa. Using the combined approach of PAI-1 gene deletion and pharmacological inhibition, we found that the release of PAI-1 in response to the exposure to P aeruginosa is an important contributor to the early alterations in the lung fluid balance caused by P aeruginosa. Moreover, PAI-1 inhibition also protected against P aeruginosa-mediated lung cell endothelial leak in vitro, indicating a direct effect of PAI-1 released by P aeruginosa on the barrier function of lung endothelial cells via a RhoA-dependent mechanism.
The importance of RhoA in mediating the inflammatory response elicited by P aeruginosa is supported by our previous in vivo and in vitro studies that reported a central role for this small GTPase in the regulation of the endothelial barrier permeability after P aeruginosa challenge.15 19 There are several pathways that could lead to RhoA activation by P aeruginosa in pulmonary endothelial cells that include the activation of TLR4 and TLR5 by the bacterial endotoxin and flagellin.23 24 Our results indicate that RhoA activation and PAI-1 expression induced by P aeruginosa depend in part on a TLR4-mediated phosphorylation of the p38 MAP kinase and activation of RhoA. However, P aeruginosa may also activate RhoA signalling via the exotoxins S and T from the type III secretion system.25 Interestingly, a recent study from Dr Lynch's laboratory has demonstrated cooperation between the endotoxin O-antigen and the type III secretion system in both laboratory and clinical isolates of P aeruginosa.26 These investigators found that the loss of O-antigen facilitates secretion of the toxins of the type III secretion system, documenting an association between endotoxin O-antigen structural composition and type III cytotoxin secretion in a mouse model of P aeruginosa pneumonia.26 These results indicate that the activation of RhoA signalling by P aeruginosa is a critical step both for the induction of the PAI-1 gene and protein and for the modulation of lung vascular permeability by this bacterium.
Since there are no known intracellular functions reported for PAI-1, we next hypothesised that PAI-1 could exert its effects from the extracellular milieu as the extracellular release of PAI-1 by lung endothelial cells was also increased after exposure to P aeruginosa. We observed that human recombinant PAI-1 increased RhoA, but decreased Rac-1 activity in these cell monolayers. Interestingly, we reported a similar shift in the balance between RhoA and Rac-1 activity in a previous study 15 where lung endothelial cell monolayers were simply exposed to P aeruginosa, indicating that the effect of P aeruginosa on the endothelial barrier permeability may depend in part on PAI-1-mediated RhoA activation. Although the mechanism(s) of small GTPase regulation by extracellular PAI-1 remain largely unknown, PAI-1 interaction with the extracellular matrix may explain in part the activation of RhoA by this coagulation protein. Indeed, PAI-1 prevents the binding of αv-integrins to the RGD domain of vitronectin, their extracellular ligand.27 Under steady-state conditions, some members of the αv-integrin family, such as the αvβ3 integrin, are anchored to the cortical actin and are associated with other cell adhesion protein complexes within the basal membrane called focal adhesion complexes. The spatial association of these proteins is under the control of local membrane activity of various kinases, such as the focal adhesion kinase and src. Among these kinases, src has been reported to cause a sustained activation of Rac-1 through Cdc-42, allowing the maintenance of cortical actin.28 It is therefore likely that the destabilisation of this adhesion complex by PAI-1 competition with αvβ3 integrin for vitronectin may affect the basal Rac-1 activity in these cells and promote the formation of actin stress fibres through RhoA activation. Taken together, these results indicate that PAI-1 amplifies P aeruginosa-mediated RhoA activation and may play an important role in the increase of lung vascular permeability associated with P aeruginosa pneumonia.
We next sought to determine if P aeruginosa increases PAI-1 release by the alveolar epithelium. Our initial in vivo results indicate that there is a time-dependent increase in PAI-1 protein released in the distal airspace in mice with P aeruginosa pneumonia. Furthermore, clinical studies have reported that BAL fluid levels of PAI-1 are higher in patients with P aeruginosa-induced VAP than those found in other infections,9 and increased BAL levels of PAI-1 were associated with increased mortality in a cohort of intensive care patients with P aeruginosa VAP.10 Here, we found that exposure of rat alveolar type II epithelial cells to P aeruginosa also resulted in an increased PAI-1 protein expression by these cells. As observed for lung endothelial cells, the induction of PAI-1 expression by P aeruginosa in rat alveolar type II cells was also RhoA dependent, underscoring the central role of the small GTPase RhoA in the control of PAI-1 expression in the lung after exposure to this bacterium. These results are also significant because we have previously reported an important role for TGFβ1, a major inducer of PAI-1,29 in mediating the breakdown of the alveolar epithelial barrier associated with ALI.20 TGFβ1 also mediates the increase in protein permeability and inhibition of baseline and cAMP-dependent vectorial fluid transport across the alveolar epithelial barrier in experimental rodent models of ALI.20 30 In addition, P aeruginosa increases protein permeability across rat alveolar epithelial type II cell monolayers, an effect that was inhibited by a specific inhibitor of RhoA or a soluble type II receptor for TGFβ1.19 Interestingly, one of the major mechanisms mediating TGFβ1 activation in the distal lung epithelium is the activation of RhoA by interleukin 1β or thrombin agonists that results in an αvβ6-integrin-mediated activation of TGFβ1 on alveolar epithelial cells,20 suggesting that PAI-1 could activate TGFβ1 signalling via a RhoA-dependent mechanism. These results indicate that P aeruginosa causes the expression of PAI-1 protein by the distal lung epithelium and that the release of PAI-1 in the distal airspace may contribute to the breakdown of the alveolar epithelial barrier associated with P aeruginosa pneumonia.
Conflicting results have been reported concerning the overall role of PAI-1 in various experimental models of pneumonia.11 12 For example, Renckens et al have reported that PAI-1 is protective during severe Gram-negative pneumonia caused by Klebsiella pneumoniae, but did not influence the outcome of Gram-positive pneumonia caused by Streptococcus pneumoniae in mice.12 We found that PAI-1 plays an important role in bacterial clearance from the lung and survival in our experimental model of P aeruginosa pneumonia because deletion or inhibition of this protein caused a decreased neutrophil recruitment into the lung in response to the airspace instillation of P aeruginosa. Our results are in accordance with previous studies that showed that PAI-1 regulates neutrophil recruitment into several organs, such as the lung and kidney.11 31 In addition to its role in recruiting neutrophils into the lung, PAI-1 expression on the non-apoptotic neutrophil plasma membrane protects these immune cells against efferocytosis (phagocytosis of viable cells) by alveolar macrophages.32 These results indicate that PAI-1 is required for the activation of the innate immune response necessary for the eradication of P aeruginosa from the distal airspaces of the lung.
In summary, we have demonstrated a dual role for PAI-1 during the course of P aeruginosa pneumonia in mice. Despite an early protective effect of the function of the alveolar capillary barrier, the lack of PAI-1 activity was later associated with the inability to clear the bacteria from the airspace and increased mortality. This apparent dichotomy may in fact reflect a synergistic host response to bacterial lung infection that has to be tightly controlled. Indeed, an exaggerated release of PAI-1 in the extracellular space may lead to the development of severe pulmonary oedema that would overwhelm PAI-1's beneficial effects on the lung innate immune response and explain the clinical correlation between high BAL fluid and plasma levels of PAI-1 and poor outcome in patients with P aeruginosa pneumonia.10 Additional studies that will include multiple time points and several strains of P aeruginosa will be needed to answer this critical question. Despite this limitation, the present study demonstrates that PAI-1 controls the development of the early lung inflammation that is required for the activation of the innate immune response necessary for the eradication of P aeruginosa from the distal airspaces of the lung, suggesting that therapeutic approaches that would inhibit the early inflammation may be deleterious for the host. Indeed, patients with high P aeruginosa burden in their lungs, but with a weak inflammatory response, have an increased mortality from P aeruginosa pneumonia.33
References
Supplementary materials
Web Only Data thx.2010.155788
Files in this Data Supplement:
Footnotes
↵* AG, ML, and YS contributed equally to the manuscript.
Funding NIH grant GM086416 (to JFP), Belgian American Educational Foundation postdoctoral fellowship (to AG), Academic Senate Individual Investigator Grant, UCSF (to YS).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.