Article Text

Abstract
Background Chronic obstructive pulmonary disease (COPD) is characterised by oxidative stress and increased risk of lung carcinoma. Oxidative stress causes DNA damage which can be repaired by DNA-dependent protein kinase complex.
Objectives To investigate DNA damage/repair balance and DNA-dependent protein kinase complex in COPD lung and in an animal model of smoking-induced lung damage and to evaluate the effects of oxidative stress on Ku expression and function in human bronchial epithelial cells.
Methods Protein expression was quantified using immunohistochemistry and/or western blotting. DNA damage/repair was measured using colorimetric assays.
Results 8-OH-dG, a marker of oxidant-induced DNA damage, was statistically significantly increased in the peripheral lung of smokers (with and without COPD) compared with non-smokers, while the number of apurinic/apyrimidinic (AP) sites (DNA damage and repair) was increased in smokers compared with non-smokers (p=0.0012) and patients with COPD (p<0.0148). Nuclear expression of Ku86, but not of DNA-PKcs, phospho-DNA-PKcs, Ku70 or γ-H2AFX, was reduced in bronchiolar epithelial cells from patients with COPD compared with normal smokers and non-smokers (p<0.039). Loss of Ku86 expression was also observed in a smoking mouse model (p<0.012) and prevented by antioxidants. Oxidants reduced (p<0.0112) Ku86 expression in human bronchial epithelial cells and Ku86 knock down modified AP sites in response to oxidative stress.
Conclusions Ineffective DNA repair rather than strand breakage per se accounts for the reduced AP sites observed in COPD and this is correlated with a selective decrease of the expression of Ku86 in the bronchiolar epithelium. DNA damage/repair imbalance may contribute to increased risk of lung carcinoma in COPD.
- COPD
- lung cancer
- DNA repair
- Ku86
- DNA-PK
- COPD pathology
- oxidative stress
Statistics from Altmetric.com
Key messages
What is the key question?
Is the balance between DNA damage and repair altered in COPD lung?
What is the bottom line?
In COPD lung there is an unbalanced oxidant-induced DNA damage and repair correlated with a selective decrease of the expression of Ku86 in the bronchiolar epithelium.
Why read on?
Because it provides a molecular mechanism to explain the increased risk of lung cancer in patients with COPD, in addition to their smoking habit.
Introduction
Lung cancer remains a major worldwide health problem, accounting for more than a sixth of cancer deaths. The incidence of lung cancer is highest in the more developed countries of North America and northern Europe. In these regions, more than 90% of cases in men and 80% of cases in women are related to smoking.1
Cigarette smoking is also the most important risk factor for chronic obstructive pulmonary disease (COPD).2 Thus, lung carcinoma and COPD share a common risk factor, tobacco smoking, through which they also possibly share similar pathogenetic mechanisms that are still largely unknown at the molecular level.
Furthermore, COPD is an independent risk factor for lung carcinoma among smokers; increasing the risk of lung cancer twofold, with the risk increasing with COPD severity.3–5
COPD is also associated with increased oxidative stress in the lower airways which can cause DNA damage and lung carcinogenesis.6 Damage to DNA induces several cellular responses that enable cells either to eliminate or cope with the damage or to activate programmed cell death, thereby eliminating cells with potentially harmful mutations.7
DNA repair mechanisms include direct repair, base excision repair, nucleotide excision repair, double-strand break (DSB) repair, and cross-link repair.7 Efficient repair of DNA DSBs is essential for the maintenance of chromosomal integrity.8 9 DSBs are repaired either by homologous recombination or by non-homologous end-joining (NHEJ) with the latter being the primary pathway involved.8 9 Six distinct proteins function in the NHEJ pathway (Ku70, Ku86, XRCC4, DNA ligase IV, Artemis, DNA-PKcs).8 9
The heterotrimeric DNA-dependent protein kinase (DNA-PK) complex is a serine-threonine kinase activated by the presence of DSBs in DNA and composed of a catalytic subunit (DNA-PKcs) and the DNA-binding heterodimer of the regulatory subunits Ku proteins (Ku70 and Ku86). Autophosphorylation of DNA-PKcs (phospho-DNA-PKcs (pS2612)) correlates with loss of protein kinase activity and dissociation of the DNA-PKcs-Ku complex. DNA-PK is localised in the nucleus and it is critical during NHEJ because it initially recognises and binds to the damaged DNA and then targets the other repair activities to the site of DNA damage.8–10
In addition to the regulatory function of the Ku proteins in DNA-PK, heterodimers of both Ku70 and Ku86 also have independent DNA repair functions.8–10 It has been documented in vitro that oxidative stress can induce nuclear loss of Ku proteins in pancreatic epithelial cells.11
Apurinic/apyrimidinic (AP) sites are common lesions in DNA and are formed either spontaneously or as intermediates during the course of base excision repair of oxidised, deaminated and alkylated bases.12 It has been estimated that about 2×105 base lesions are generated per cell per day. The level of AP sites in cells is thought to be a good indicator of both ongoing DNA damage and repair induced by chemical agents and cell ageing.12
8-Hydroxy-2-deoxyguanosine (8-OH-dG) is produced by the oxidative damage of DNA by reactive oxygen and nitrogen species and serves as an established marker of oxidative stress.13 8-OH-dG is increased in the peripheral lung of patients with pulmonary emphysema.14 After a DNA DSB a variant histone H2A, termed H2A.x (which replaces conventional H2A in a subset of nucleosomes), is rapidly phosphorylated at Serine 139 by ATM kinase. Phospho-S139-H2A.x is also known as γ-H2AFX.15 This phosphorylation step can extend up to several thousand nucleosomes from the actual site of the DSB and may mark the surrounding chromatin for recruitment of proteins required for DNA damage signalling and repair.15 Both 8-OH-dG and γ-H2AFX are considered sensors of oxidative stress-induced DNA damage.
The aim of this study was to investigate the degree of oxidant-induced DNA damage and DNA repair and the localisation and expression of DNA-PKcs, DNA-PKcs (pS2612), Ku70, Ku86 and γ-H2AFX in lung parenchyma of patients with COPD compared with smokers with normal lung function and non-smokers (control groups). The study also analysed the effects of oxidative stress on Ku protein expression in primary human bronchial epithelial cells in vitro, the functional effect of Ku86 knockdown in bronchial epithelial cells and the effect of cigarette smoke exposure on bronchial epithelial cell Ku86 expression in an animal model in vivo.
Methods
Participants
We recruited a total of 69 patients (64 undergoing lung resection for a solitary peripheral carcinoma (11 non-smokers, 28 smokers with normal lung function and 25 with COPD) and five non-smokers undergoing lung transplantation). All patients were recruited from the Respiratory Diseases Clinic of the University Hospital of Ferrara, Italy and from the Royal Brompton Hospital, Imperial College London, UK.
For the DNA damage/repair and DNA fragility studies, 11 were smokers with COPD, 15 were smokers with normal lung function and seven were non-smokers (table 1). Samples from 14 smokers with COPD, 13 smokers with normal lung function and nine non-smokers were used for immunohistochemical and western blotting studies (table 2). COPD was defined according to international guidelines.2 All patients were stable at the time of the study and were free from acute exacerbations of symptoms and from upper respiratory tract infections in the 2 months preceding the study. None had received glucocorticoids, theophylline, antibiotics or antioxidants within the month preceding surgery or bronchodilators within the previous 48 h. Patients had no history of asthma or other allergic diseases. Former smokers had stopped smoking for more than 1 year. Each patient underwent medical history, physical examination, chest radiography, electrocardiogram, routine blood tests, and pulmonary function tests during the week prior to surgery. The study was approved by the ethics committees of the University Hospital of Ferrara and the Royal Brompton Hospital.
Characteristics of participants taking part in the DNA damage/repair and DNA fragility studies
Characteristics of participants taking part in the immunohistochemistry and western blotting studies
Pulmonary function tests were performed as previously described16 according to published guidelines.17 Predicted values for the different measures were calculated from the regression equations published by Quanjer and colleagues.17
Experimental methods
The cigarette smoke-exposed mouse model, the immunohistochemistry in human and mouse lungs for DNA-PKcs, DNA-PKcs (pS2612), Ku70, Ku86 and γ-H2AFX, the DNA damage/repair quantification and DNA fragility assays, the western blot analysis for Ku70 and Ku86 in human lung parenchyma, the culture of primary human bronchial epithelial cells and the Ku86 knock-down siRNA in human bronchial epithelial cells are all described in full detail in the online data repository.
Statistical analysis
Group data were expressed as mean and SD or median (25–75 percentiles), as appropriate. Analysis of variance for clinical data and the Kruskal–Wallis test for histological data were used to determine differences between groups. The Mann–Whitney U test was performed after the Kruskal–Wallis test when appropriate. Bonferroni adjustment was applied when indicated. Categorical values were compared by means of the χ2 test. A probability value of <0.05 was considered statistically significant.
Results
Clinical data
Tables 1 and 2 show the characteristics of the patients recruited. There was no significant difference in the smoking history between patients with COPD and smokers with normal lung function (p=0.8279 and p=0.5369 for pack-years of COPD vs smokers in tables 1 and 2). The prevalence of chronic bronchitis was 52% in the COPD group and 50% in the smokers with normal lung function.
DNA damage/repair in lung parenchyma
DNA samples from patients with COPD were more fragile and liable to degradation than those obtained from non-smokers and smokers with normal lung function (figure 1A). In order to explain this we examined markers of specific DNA damage and repair. The degree of oxidative stress-induced DNA damage was similar in healthy smokers and smokers with COPD as determined by the level of 8-OH-dG/μg DNA (figure 1B) and statistically significantly greater than that observed in normal controls (0.26 (0.17) and 0.29 (0.17) vs 0.06 (0.04) respectively; p=0.0033). However, the number of AP sites, a marker of DNA damage and repair, was elevated in smokers with normal lung function (2.863 (1.31)) compared with healthy non-smokers (1.195 (0.33); p=0.0012) and patients with COPD (1.607 (0.73); p=0.0148). The number of AP sites in patients with COPD, however, was not statistically significantly different compared with non-smokers (p=0.2048) (figure 1C). The current smoking status had no effect on AP sites.

(A) Ethidium bromide staining of freeze-thawed DNA extracted from non-smokers (n=6), smokers with normal lung function (n=6) and patients with mild to moderate COPD (n=10). (B) Graphical representation of 8-hydroxy-2-deoxyGuanosine (8-OH-dG) content, an index of oxidative stress-induced DNA damage, in human lung parenchyma obtained from non-smokers (n=7), healthy smokers with normal lung function (n=15) and patients with COPD (n=11). Histograms represent mean (SD). (C) Graphical representation of AP sites, indexes of DNA damage and repair, in human lung parenchyma obtained from non-smokers (n=7), healthy smokers with normal lung function (n=15) and patients with COPD (n=11). Histograms represent mean (SD).
Immunohistochemistry for DNA-PKcs, DNA-PKcs (pS2612), Ku70, Ku86 and γ-H2AFX in human lung parenchyma
The authors postulated that there may be decreased DNA repair in COPD, therefore components of this process were analysed. Ku86 was mainly localised to the nucleus in both structural and inflammatory cells of the lungs (figure 2). The number of bronchiolar epithelial cells without nuclei positively stained for Ku86 was statistically significantly higher in patients with COPD (median (IQR) 3.54% (1.85–5.75), n=14), but not in healthy smokers (1.5% (0.3–5.4), n=13), compared with non-smokers (1% (0.6–1.8), n=9) (p<0.039). In addition, the amount of Ku86 protein (0.26 (0.04) vs 0.4 (0.03) Ku86/actin ratio OD arbitrary units; p=0.026), but not total Ku70 (0.49 (0.14) vs 0.4 5(0.1); p=0.82) in lung parenchyma demonstrated a statistically significantly decrease in COPD (n=6) compared with smokers with normal lung function (n=6) (figure 2D).
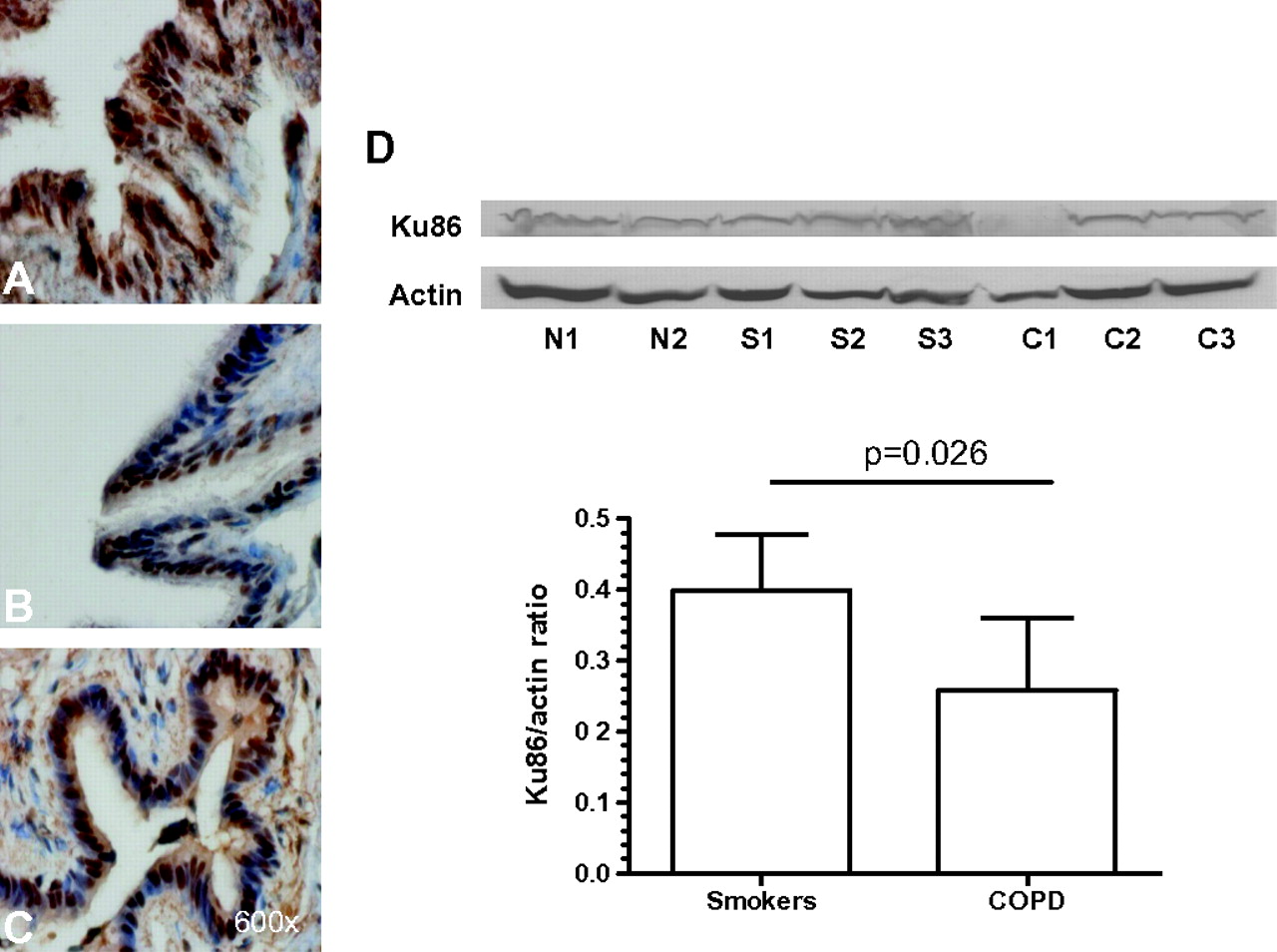
Photomicrographs showing the bronchiolar epithelium from a non-smoker (A), a healthy smoker with normal lung function (B) and a patient with mild to moderate chronic obstructive pulmonary disease (COPD) (C) immunostained for identification of Ku86+cells (brown). Results are representative of those from nine non-smokers, 13 smokers with normal lung function, 14 patients with mild/moderate COPD. (D) Western blotting analysis of Ku86 (with actin controls) in whole lung parenchyma lysates from one non-smoker (N), three healthy smokers with normal lung function (S) and three patients with mild to moderate COPD (C). Histograms represent the mean (SD) of the Ku86/actin ratio in healthy smokers (n=6) and patients with COPD (n=6).
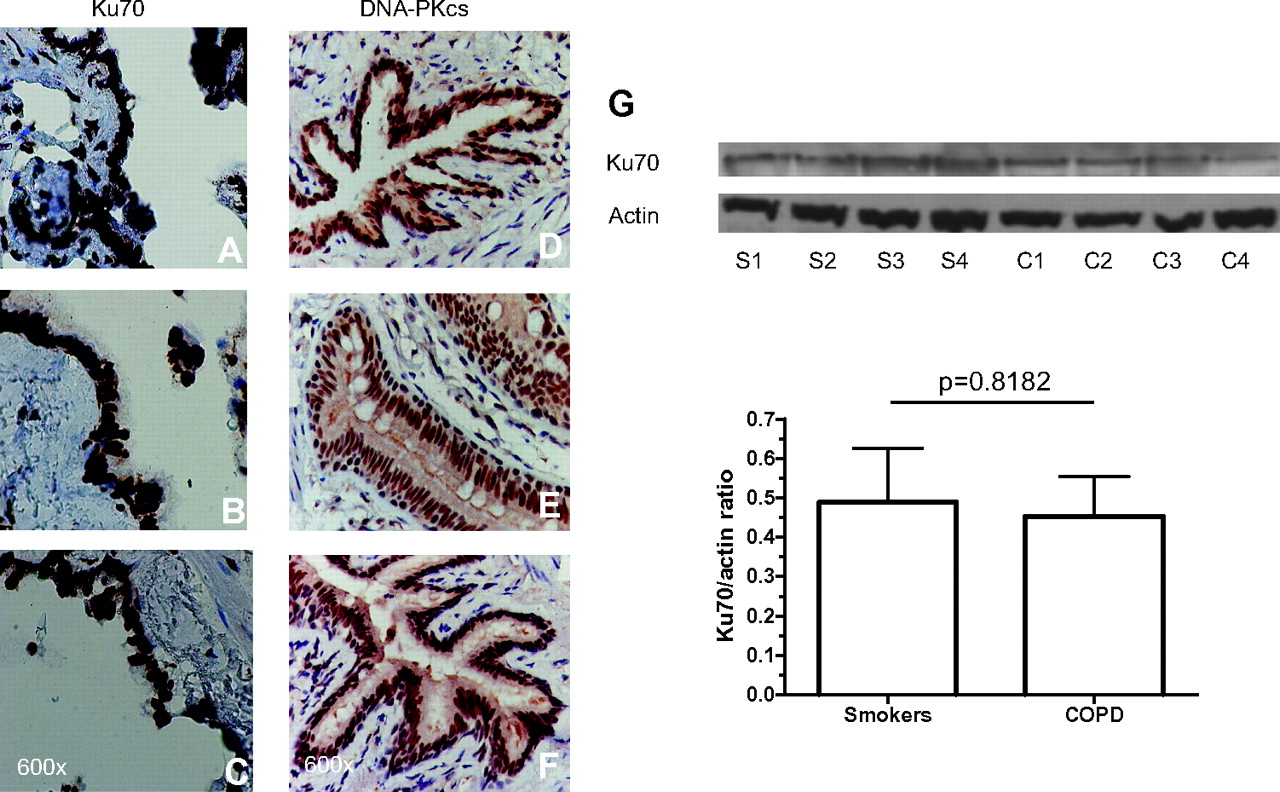
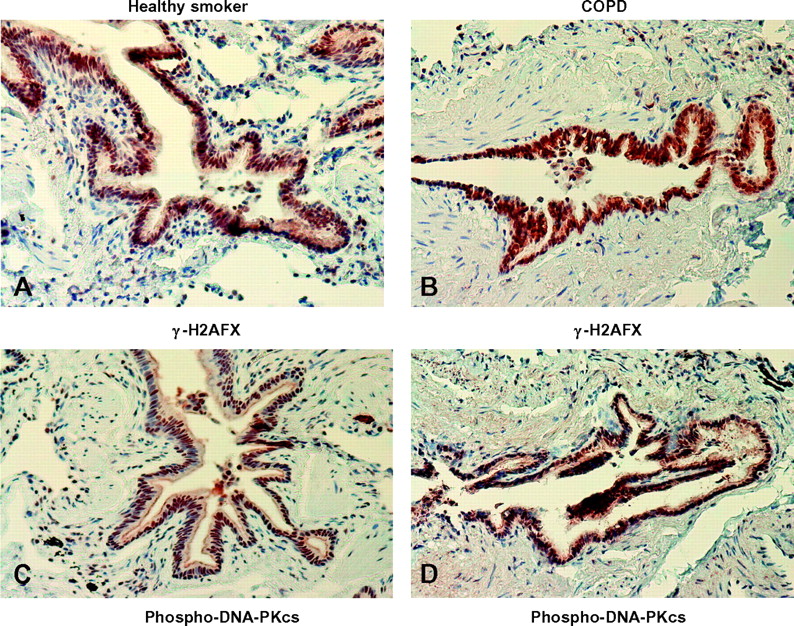
DNA-PKcs, DNA-PKcs (pS2612), Ku70 and γ-H2AFX were localised exclusively in the nucleus in all cells, including bronchiolar epithelial cells (all bronchiolar epithelial cells were stained in all three groups). There was no statistically significant difference in their localisation and global expression among the three groups for DNA-PKcs and Ku70 (figures 3 and 4 and online table S1) and between smokers with normal lung function and patients with COPD for DNA-PKcs (pS2612) and γ-H2AFX (figures 3 and 4 and online table S1).

Photomicrographs showing the bronchiolar epithelium from a non-smoker (A, D), a healthy smoker with normal lung function (B, E) and a patient with mild to moderate chronic obstructive pulmonary disease (COPD) (C, F) immunostained brown for identification of Ku70+ (left panel) and DNA-PKcs+cells (right panel). Results are representative of those from nine non-smokers, 13 smokers with normal lung function and 14 patients with mild to moderate COPD. (G) Western blotting analysis of Ku70 (with actin controls) in whole lung parenchyma lysates from four healthy smokers with normal lung function (S) and four patients with mild to moderate COPD (C). Histograms represent the mean (SD) of the Ku70/actin ratio in healthy smokers (n=6) and COPD (n=6).

Photomicrographs showing the bronchial epithelium from a healthy smoker with normal lung function (A, C) and a patient with mild to moderate chronic obstructive pulmonary disease (COPD) (B, D) immunostained for identification of γ-H2AFX+(A, B) and phospho-DNA-PKcs+(C, D) cells (brown). Results are representative of those from 13 healthy smokers with normal lung function and 14 patients with mild to moderate COPD.
Furthermore, no statistically significant differences in the percentage of endoalveolar macrophages stained for DNA-PKcs, Ku70, and Ku86 among the three groups and between smokers with normal lung function and patients with COPD for DNA-PKcs (pS2612) and γ-H2AFX were seen (all endoalveolar macrophages were stained in all three groups).
Immunohistochemistry for Ku70 and Ku86 in mouse lung parenchyma
Using a smoke-exposed mouse model the authors were also able to demonstrate predominant nuclear Ku 70 and Ku86 localisation in bronchial epithelial cells (figure 5). The expression of Ku86 in the bronchial epithelial cells was decreased to a statistically significant extent only in susceptible A/J mice (p=0.012) but not in C57BL/6J mice (p=0.44) exposed to cigarette smoke. This decrease was prevented by treatment of A/J mice with N-acetylcysteine (NAC), as determined by immunohistochemistry (A/J mice: median (interquartile range) 75% (71%–83%) for controls vs 66% (57%–70%) for smoke-exposed mice vs 73% (67%–78%) for NAC+smoke-exposed mice; p=0.012 for controls vs smoke-exposed mice; C57BL/6J: 75% (68%–88%) for controls vs 71% (65%–83%) for smoke-exposed mice; p=0.44) (figure 5). In contrast, the expression of Ku70 in the bronchial epithelial cells was not statistically significantly different between controls and smoke-exposed mice in either A/J or C57BL/6J strains (data not shown).

Photomicrographs showing the bronchial epithelium from a control (A, D) and a cigarette-smoke exposed (B, E) mouse respectively of the C57BL/6J (A, B) and A/J (D, E) strains immunostained for identification of Ku86+cells (brown). Histograms represent the median (IQR) of the percentage of bronchial epithelial cells with nuclear immunostaining for Ku86 on total number of bronchial epithelial cells in controls (n=6 and 8 for C57BL/6J and A/J strains respectively) and cigarette-smoke exposed mice (n=7 and 11 for C57BL/6J and A/J strains respectively) of C57BL/6J (C) and A/J (F) strains.
Effect of H2O2 on Ku70/Ku86 expression in primary human bronchial epithelial cells
Stimulation of primary human bronchial epithelial cells with hydrogen peroxide in vitro produced a statistically significant reduction in Ku86 but not Ku70 expression (p<0.0112; figure 6A,B). Additionally, knockdown of Ku86 protein in bronchial epithelial BEAS-2B cells using siRNA resulted in a statistically significant reduction of the ability of these cells to induce AP sites in response to H2O2 exposure (figure 6C–E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graphical representation of Ku86 (A) and Ku70 (B) expression, measured by immunoenzymatic analysis, in nuclear extracts from primary human bronchial epithelial cells before (control) and after stimulation with hydrogen peroxide (H2O2). (C) Real time PCR analysis of Ku86 mRNA expression in lysates from wild type (Wt) and Ku86 knockdown (KD) BEAS-2B cells. (D) Western blotting analysis of Ku86 expression in lysates from Wt and KD BEAS-2B cells. (E) Effects of Ku86 knockdown in bronchial epithelial BEAS-2B cells using siRNA in the modulation of the ability of these cells to induce AP sites in response to hydrogen peroxide stimulation. Results are expressed as mean (SD) and are representative of three separate experiments. *p compared with control cells at t=0 h; §p compared with Ku86 siRNA-treated cells at t=12 h.
Discussion
There is a selective decrease in the level of the DNA repair protein Ku86 in the bronchiolar epithelium of patients with COPD. This has also been confirmed in the cigarette-smoke exposed murine strain susceptible to lung cancer. In vitro, the exposure of bronchial epithelial cells to H2O2 reproduced the selective pattern of Ku86 expression decrease observed in COPD. In addition, the study demonstrated for the first time that the number of AP sites is significantly increased in lung parenchyma from smokers with normal lung function compared with non-smokers, whereas the level of 8-OH-dG is significantly increased in lung parenchyma from all smokers with or without COPD compared with non-smokers.
AP levels reflect both ongoing DNA damage and DNA repair of the damage. To the authors' surprise, this combined marker of DNA damage/repair was higher in smokers with normal lung function compared with smokers with COPD. In contrast, 8-OH-dG and γ-H2AFX, markers of DNA damage, were not significantly different between smokers with normal lung function and current and former smokers with COPD, reflecting similar levels of smoking exposure. The absence of increased AP levels in patients with COPD might be a consequence of a loss of DNA repair secondary to a lack of Ku86 in bronchiolar epithelial cells.
Furthermore, short-term (11 days) cigarette smoke exposure in mice decreased K86 expression in bronchial epithelial cells only in the A/J strains, susceptible to tobacco-smoke induced lung cancer, and this effect is prevented by the treatment with the antioxidant NAC. Finally, in vitro studies demonstrated that in bronchial epithelial cells H2O2 can decrease Ku86, but not Ku70, expression, and Ku86 knockdown demonstrates a significant modulation of AP levels by preventing restoration of levels to baseline.
A previous study demonstrated a reduction of DNA-PK activity in peripheral blood mononuclear cells (PBMCs) obtained from patients with non-small cell lung cancer (NSCLC) compared with those without lung cancer.18 The same study also demonstrated that the DNA-PK activity in PBMCs reflected that seen in bronchial epithelial cells.18 This reduced DNA-PK activity may be linked to the decreased Ku86 expression reported here. Several studies have already shown that despite leucocytes not being the direct target of tobacco carcinogens, the level of DNA damage in PBMCs correlates with tobacco smoking-induced DNA damage in human lung tissues.19 20 This is in keeping with the concept of COPD having a major systemic component.21
The absence of significant differences in bronchiolar expression of phospho-DNA-PKcs in this study may suggest that DNA-PKcs autophosphorylation, which in vitro correlates with loss of protein kinase activity and dissociation of the DNA-PKcs-Ku complex,9 may not be an important mechanism in COPD lungs.
COPD is an independent risk factor for lung carcinoma, particularly for squamous cell carcinoma, among smokers.4 It is also associated with increased oxidative stress which can cause DNA damage.6 Lung cancer is up to five times more likely to occur in smokers with airflow obstruction than those with normal lung function5 and lung cancer is one of the commonest causes of death among patients with COPD.22 The authors hypothesise that a defective DNA repair mechanism in smokers with COPD in response to oxidative stress will contribute to the increased incidence of lung carcinoma in these patients.
Recent evidence has determined a critical role for acetylation of histone H3 on lysine 56 (AcH3K56) in DNA damage and repair.23 24 Furthermore, deacetylation of AcH3K56 is controlled by histone deacetylases (HDACs) 1 and 2 and loss of HDAC1 and/or 2 can increase susceptibility to UVA irradiation.23 Further evidence for a role of HDAC-containing complexes in the regulation of DNA damage and repair is seen in Caenorhabditis elegans in response to ionising radiation25 and there also appears to be an important role for sirtuins in deacetylating H3K56 in mammalian cells.24 26 This suggests that in diseases such as COPD where there is a decrease in both HDAC227 and SIRT128 expression/activity in the lungs there will be less protection against DNA strand break and repair induced by environmental factors, increasing the potential for somatic mutations and increasing the risk of lung cancer.
Ku86-deficient mice have early senescence and mortality but do not show an increased incidence of lung cancer.29 However, the effects of long-term tobacco smoke exposure on these animals have not been investigated. Further studies using a conditional Ku86 knockout chronically exposed to tobacco smoke will aid the understanding of the role of decreased Ku86 in facilitating lung cancer.
In conclusion, these data suggest that a reduced expression of the DNA repair protein Ku86 in the nucleus of the bronchiolar epithelial cells from patients with COPD can contribute to their increased risk of developing lung carcinoma.
References
Supplementary materials
Web Only Data thx.2010.156448
Files in this Data Supplement:
Web Only Data thx.2010.156448
Files in this Data Supplement:
Footnotes
Gaetano Caramori, Ian M Adcock, Peter J Barnes and Alberto Papi have contributed equally to this work.
Funding The study was supported by grants from Fondazione Carife (Ferrara, Italy; http://www.fondazionecarife.it/) and Associazione per la Ricerca e la Cura dell'Asma (ARCA, Padova, Italy) and the Wellcome Trust.
Competing interests None.
Ethics approval This study was conducted with the approval of the Ethics committees of the University Hospital of Ferrara and the Royal Brompton Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.