Article Text

Abstract
Background Thymic stromal lymphopoietin (TSLP) is an epithelial cell-derived cytokine that strongly activates dendritic cells and can initiate allergic inflammation. Since exposure to rhinovirus or double-stranded (ds) RNA (a surrogate of viral infection) induces TSLP expression in bronchial epithelial cells (BECs), this cytokine may link innate antiviral responses and the type 2 adaptive immune response.
Objective As BECs from donors with asthma have a deficient interferon (IFN) response to rhinovirus infection, a study was undertaken to test the hypothesis that their antiviral response shows a bias towards TSLP production.
Methods Primary BECs were grown from subjects with asthma and healthy volunteers. After exposure to dsRNA, interleukin (IL)-8, IFNβ and TSLP mRNA and protein expression were measured by RT-qPCR and ELISA, respectively.
Results dsRNA dose-dependently increased IL-8 expression in BECs with no significant difference between the groups. However, BECs from subjects with asthma expressed less IFNβ and more TSLP mRNA and protein in response to dsRNA than BECs from those without asthma (median (IQR) 57 (38–82) pg/ml vs 106 (57–214) pg/ml for IFNβ (p<0.05) and 114 (86–143) pg/ml vs 65 (32–119) pg/ml for TSLP (p<0.05) in response to 10 μg/ml dsRNA for 24 h). Induction of TSLP mRNA by dsRNA was blocked by Toll-like receptor 3 or protein kinase inhibitors or by preventing de novo protein synthesis, but not by neutralisation of type I IFN receptors.
Conclusion BECs from subjects with asthma are biased towards higher TSLP and lower IFNβ production in response to dsRNA, suggesting that viral infection in asthma may lead to an altered mediator profile that biases towards a Th2 immune response.
- Interferon
- TSLP
- rhinovirus
- asthma
- exacerbation
- epithelial cells
- airway epithelium
- asthma mechanisms
- innate immunity
Statistics from Altmetric.com
- Interferon
- TSLP
- rhinovirus
- asthma
- exacerbation
- epithelial cells
- airway epithelium
- asthma mechanisms
- innate immunity
Introduction
Exacerbations of asthma constitute a major proportion of asthma morbidity and mortality1 and account for significant healthcare costs, identifying an important unmet need. It is now evident that asthma deterioration and exacerbations in children and adults are frequently associated with rhinovirus (RV) infection,2 3 and that the bronchial epithelium is the target for the virus.4 5 Furthermore, infants who develop severe viral respiratory infections are more likely to develop asthma later in childhood,6 suggesting a potential link between infection and disease pathogenesis.
It has been shown that RV-exposed primary bronchial epithelial cells (BECs) from individuals with asthma have reduced innate antiviral responses and are more permissive for viral replication than cells from healthy controls.7 The antiviral response of BECs from subjects with asthma involved reduced generation of the type I and III interferons IFNβ7 and IFNλ,8 respectively. This deficient innate immune defence against RV infection is probably of pathogenic importance in exacerbations of asthma.7 However, despite increased viral infection of BECs from subjects with asthma, RV-induced epithelial production of cytokines such as interleukin (IL)-6 and RANTES did not differ between subjects with asthma and healthy controls.7–9
Recently, an IL-7-like cytokine, thymic stromal lymphopoietin (TSLP), has been put forward as a particularly important epithelium-derived mediator in asthma.10 11 TSLP is produced by several epithelial cell lines, keratinocytes12 and airway epithelium.13 The pathophysiology of human TSLP involves innate allergic immune responses with direct effects of TSLP on mast cells and on immature dendritic cells (DCs).14 15 TSLP-activated DCs initiate adaptive allergic immune responses by triggering differentiation of naïve T cells into inflammatory Th2 cells that produce several traditional Th2 cytokines and also large amounts of tumour necrosis factor α (TNFα).10 14–16 Importantly, Ying et al13 17 observed that bronchial epithelial expression of TSLP mRNA is increased in severe asthma.
Factors that may increase TSLP expression, including viral stimuli, have been examined in vitro using BECs from healthy human individuals.14 18 19 However, to our knowledge, no specific information is available on generation of TSLP in BECs from subjects with asthma to support the possibility of involvement of this cytokine in viral infection-evoked exacerbations of asthma. During replication, single-stranded (ss) RNA viruses such as RV produce double-stranded (ds) RNA which is detected as a ‘danger signal’ by the innate immune system. Double-stranded RNA appears to be crucial for many of the biological activities of RV infections since non-replicative RV and/or ssRNA are poor stimuli for epithelial interferon and cytokine generation.7 20 Polyinosine-polycytidylic acid (poly (I:C)) is a synthetic dsRNA that acts as a molecular pattern associated with viral infections and has been shown to mimic the effects of RV infection in terms of inflammatory regulatory molecules and unresponsiveness to glucocorticoids.21–23 Based on its utility as a surrogate of viral infection, we hypothesised that exposure of BECs from subjects with asthma to dsRNA would strongly induce TSLP expression in association with lower levels of IFNβ compared with BECs from normal subjects. We therefore investigated the expression of TSLP, IFNβ and IL-8 in response to treatment of BECs obtained from subjects with and without asthma with dsRNA.
Methods
Subjects
Two groups of subjects were recruited from the departmental database and asthma clinic. These were healthy subjects without asthma and a group of subjects with asthma ranging from mild intermittent to moderate/severe persistent disease characterised according to the Global Initiative for Asthma guidelines involving symptoms, pulmonary function and medication as described in the online supplement. Inclusion criteria were non-smokers or those with a history of <5 pack-years (no current smokers) and no upper respiratory tract infection within the last 2 months. Subjects not fulfilling these inclusion criteria and pregnant or lactating women were excluded.
Fibreoptic bronchoscopy
Epithelial brushings were obtained by bronchoscopy using a fibreoptic bronchoscope (FB-20D; Olympus, Tokyo, Japan) in accordance with standard published guidelines and bronchial brushings were obtained using a standard sterile single-sheathed nylon cytology brush (BC 9C-26101; Olympus), all as previously described and explained in more detail in the online supplement.
Epithelial cultures
Primary cultures were established by seeding bronchial brushings into collagen-coated tissue culture flasks containing 3 ml serum-free hormonally-supplemented bronchial epithelium growth medium (BEGM; Clonetics, San Diego, California, USA),24 as described in detail in the online supplement. BECs were seeded into 12-well plates (Nunc, Life Technologies) and, when 80–90% confluent, they were stimulated with poly (I:C) (Invitrogen Ltd, Paisley, UK) at different doses and time points (see online supplement).
Inhibition of TSLP mRNA expression in bronchial epithelial cells
The Toll-like receptor 3 (TLR3) inhibitor chloroquine (Sigma-Aldrich, Gillingham, UK), an imidazolo-oxindole inhibitor of dsRNA-activated protein kinase (PKR), or its negative control (5-chloro-3-(3,5-dichloro-4-hydroxybenzylidene)-1,3-dihydro-indol-2-one) (both from Merck-Calbiochem, Nottingham, UK) were added to BEC cultures 2 h prior to stimulation with 1 μg/ml dsRNA. Protein synthesis was blocked by preincubating the BECs with cycloheximide (Sigma-Aldrich) for 1 h before stimulation. A neutralising antibody for the type I IFN receptor (Interferonsource-PBL, Biomedical Laboratories, Piscataway, New Jersey, USA) and its negative control mouse IgG2a isotype control (eBioscience, Hatfield, UK) were added at a concentration of 12 μg/ml at the time of addition of dsRNA. BECs were stimulated for 3 h and processed for mRNA expression as described below.
Cytokine measurement by ELISA
Release of TSLP (R&D Systems, Abingdon, UK), IL-8 (Biosource International, California, USA) and IFNβ (PBL-Interferonsource, New Jersey, USA) into culture supernatants of BECs was measured at 24 h after stimulation with dsRNA using ELISA kits according to the manufacturers' instructions.
Quantification of IL-8, IFNβ and TSLP gene expression using reverse transcription and quantitative PCR (RT-qPCR)
Total RNA was extracted from primary BECs using TRIzol (Invitrogen, Carlsbad, California, USA) according to the manufacturer's instructions and treated with RNase-free DNase I (Ambion, Huntingdon, UK) to remove any contaminating genomic DNA. RT-qPCR was performed as described in the online supplement.
Statistical analysis
Statistical analysis was performed using the SPSS 14.0 software package for Windows (Chicago, Illinois, USA). With the exception of the studies using pharmacological inhibitors, all data are reported as median values with interquartile ranges and were analysed using non-parametric tests. Data within each group (asthma and healthy) were analysed using the Friedman test and the Wilcoxon test for two-paired samples was performed for each group. Statistical differences between asthma and healthy groups were determined using the Mann–Whitney U test for unrelated samples. For the inhibition studies, the data were normalised relative to the dsRNA-treated BEC cultures and are given as mean±SD; statistical comparisons were made using the Student t test. A p value <0.05 was considered significant.
Results
Clinical characterisation of subjects
Twenty-six subjects participated in the study, 13 healthy individuals and 13 with asthma. The clinical characteristics of the subjects are shown in table 1.
Clinical characteristics of study subjects
Effects of dsRNA on IL-8 expression
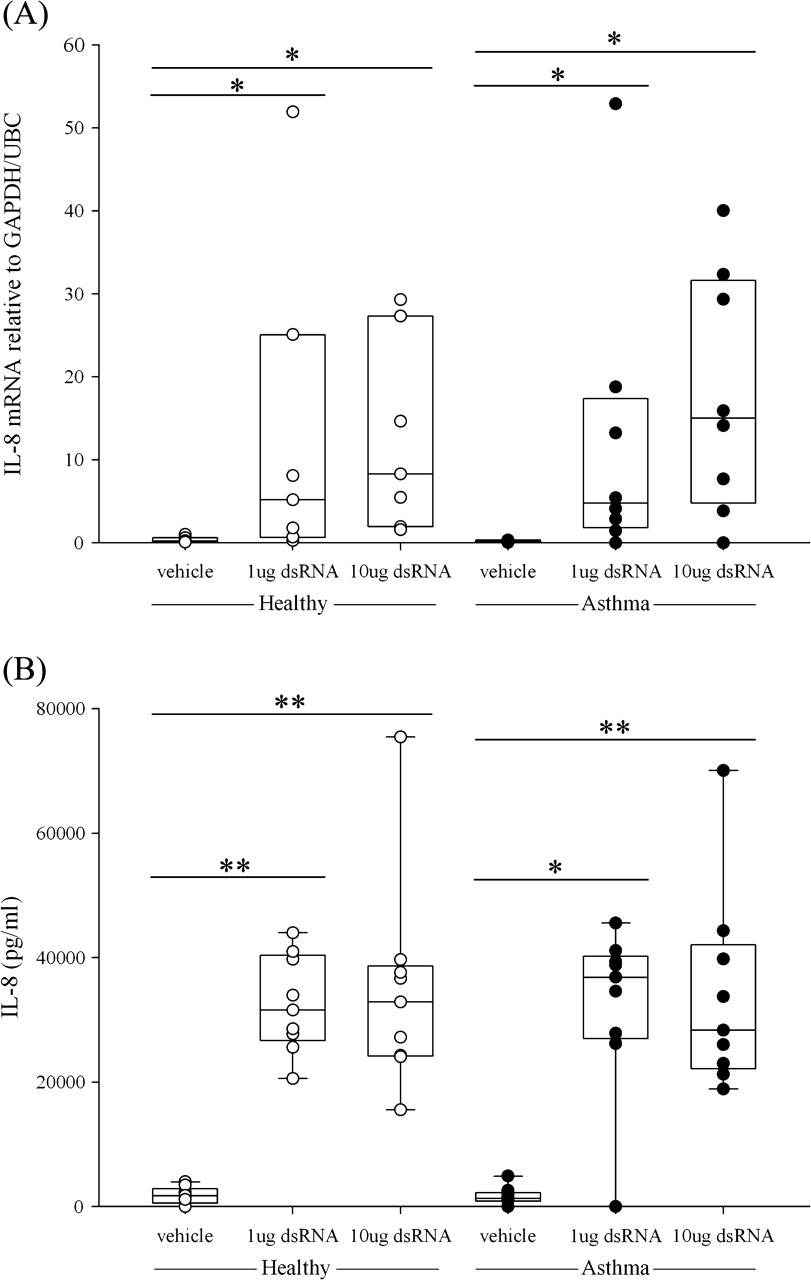
IL-8 is regarded as a cytokine important for the innate immune response and is a mediator of neutrophilic inflammation.20 Treatment of BECs with dsRNA dose-dependently increased IL-8 gene expression at 3 and 6 h after treatment and this was sustained at 24 h (figure 1A and figure E1 in online supplement). Comparing IL-8 mRNA levels between the asthma and healthy groups, there was no significant difference in gene expression. Protein levels of IL-8 were detectable in the cell supernatants, but again there was no significant difference between BECs from healthy subjects and those with asthma (figure 1B).

Induction of (A) interleukin (IL)-8 mRNA expression and (B) IL-8 protein expression in primary bronchial epithelial cells from healthy subjects (open circles) or subjects with asthma (closed circles) after stimulation with double-stranded RNA (dsRNA) at (A) 3 h and (B) 24 h. The box plots show the median and interquartile range, and the bars show the 10th and 90th percentiles. *p<0.05; **p<0.01 vs control untreated cells (Wilcoxon rank sum test).
Innate immune response to dsRNA: induction of IFNβ expression
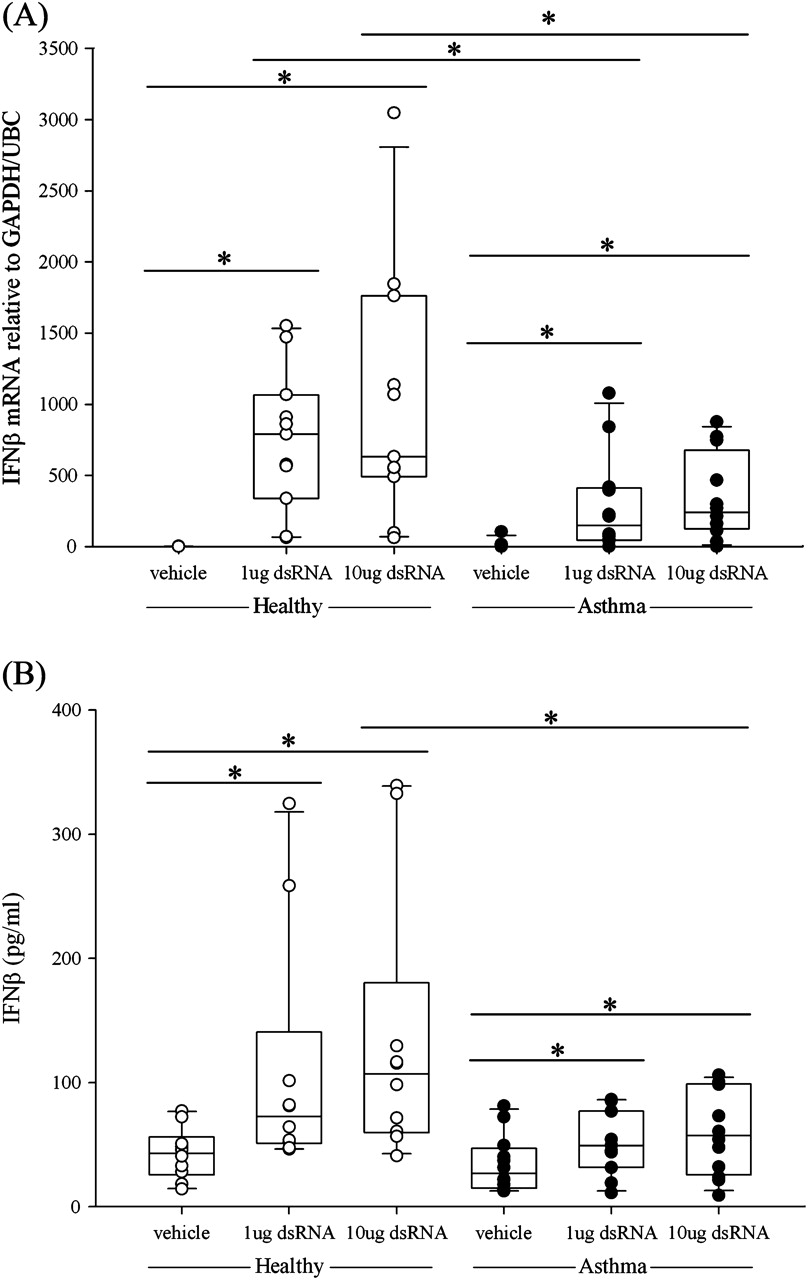
Bronchial epithelial IFNβ mRNA expression was induced at 3, 6 and 24 h after stimulation with dsRNA. In both the healthy and asthma groups, dsRNA induced a dose-dependent increase in IFNβ mRNA which was maximal at 3 h. However, the BECs from subjects with asthma expressed significantly lower levels of IFNβ mRNA than those from healthy controls (1 μg dsRNA, p<0.02; 10 μg dsRNA, p<0.04; figure 2A). At 24 h, IFNβ gene expression was still significantly increased in the healthy group and was higher than in the asthmatic group (p<0.02; figure E2 in online supplement). The lower expression of IFNβ mRNA seen in response to dsRNA in BECs from subjects with asthma was confirmed at the protein level using ELISA. In both groups, a dose-dependent increase in IFNβ protein was observed, but IFNβ protein levels were significantly lower in the asthma group (figure 2B), as previously reported for RV infection.7
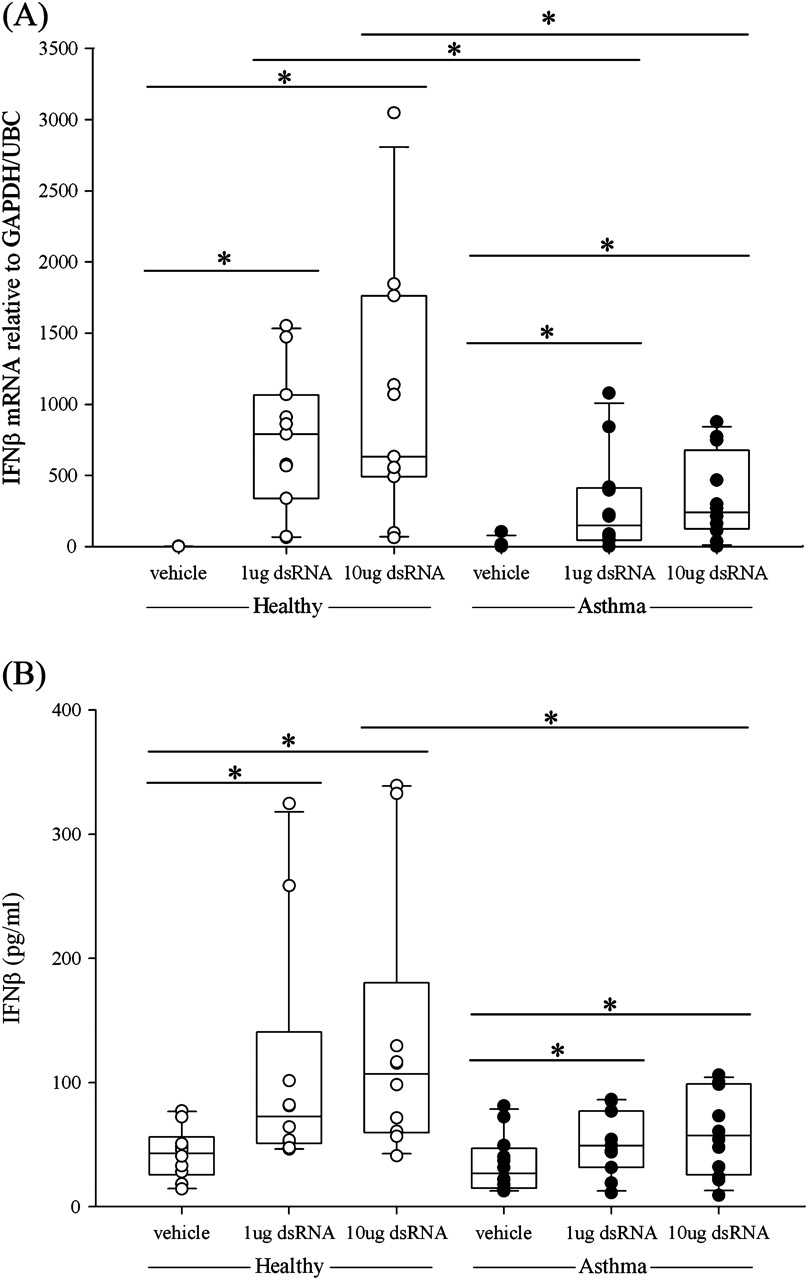
Induction of (A) interferon β (IFNβ) mRNA expression and (B) IFNβ protein release following treatment of bronchial epithelial cells from healthy subjects (open circles) or subjects with asthma (closed circles) after stimulation with double-stranded RNA (dsRNA) at (A) 3 h and (B) 24 h. The box plots show the median and interquartile range, and the bars show the 10th and 90th percentiles. *p<0.05 vs control untreated cells (Wilcoxon rank sum test). Between-group comparisons were made using the Mann–Whitney U test.
Time course and dose response of dsRNA-induced TSLP expression in BECs
Since TSLP has been reported to be rapidly induced in BECs after viral stimulation, we included two early (3 and 6 h) and two later time points (24 h and 48 h) for gene analysis with RT-qPCR and ELISA analysis for protein measurements. TSLP mRNA was dose-dependently induced at 3 h after stimulating the cells with dsRNA in both groups (figure 3A). At 6 h the asthma group maintained a significant and dose-dependent increase in TSLP mRNA while no significant (p>0.05) TSLP mRNA was expressed in the healthy group (see figure E3 in online supplement). At both 3 h and 6 h after exposure to dsRNA, the highest dose of dsRNA (10 μg/ml) produced significantly higher levels of TSLP mRNA in BECs from subjects with asthma than in BECs from healthy controls (p<0.05 and p<0.04). At the later time points, TSLP mRNA was no longer increased after dsRNA stimulation compared with baseline levels (see figure E4 in online supplement). Similarly, at 48 h no increase in TSLP mRNA was observed (data not shown). At 24 h after treatment, generation of TSLP protein by the BECs was dose-dependently increased by dsRNA in both the healthy and asthma groups (figure 3B). Similar to the gene expression data, significantly higher levels of TSLP protein were detected in the asthma group after 10 μg/ml dsRNA stimuli compared with healthy individuals (p<0.04).
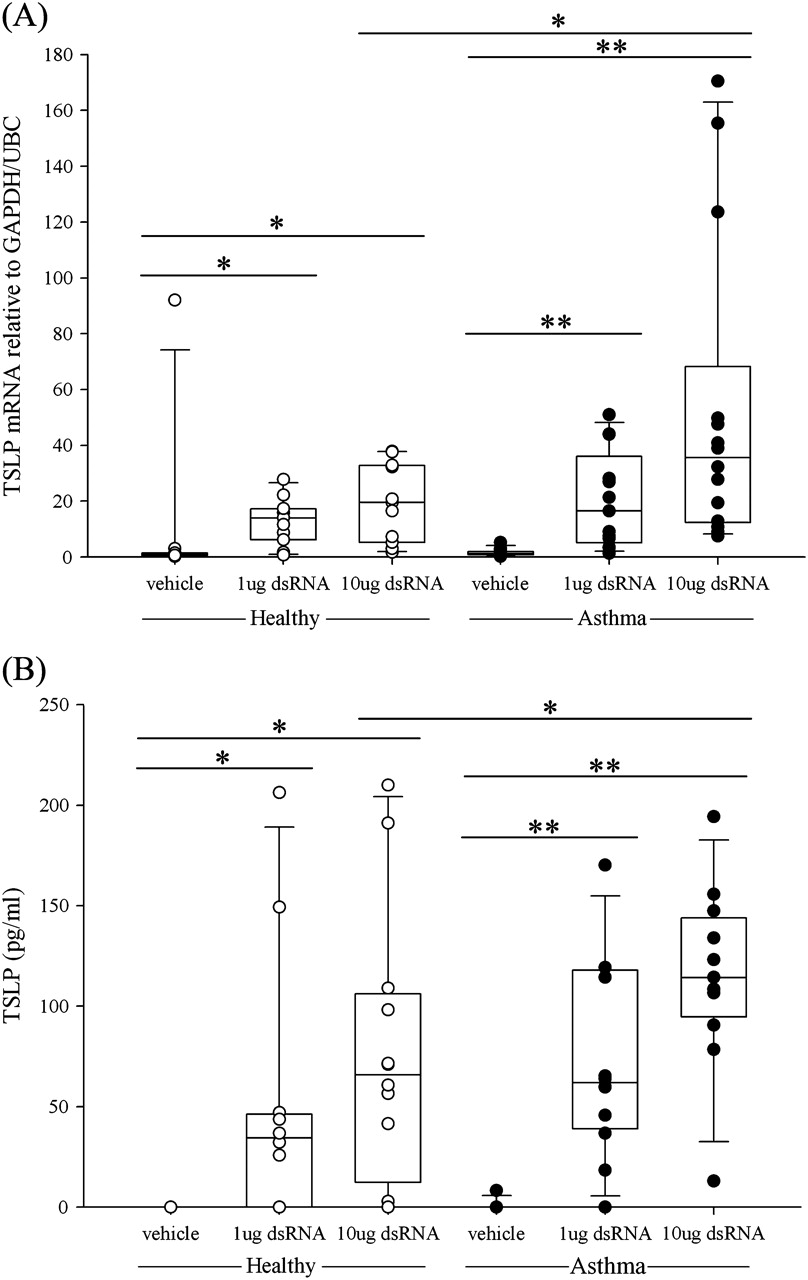
Induction of (A) thymic stromal lymphopoietin (TSLP) mRNA expression and (B) TSLP protein release in bronchial epithelial cells from healthy subjects (open circles) or subjects with asthma (closed circles) after stimulation with double-stranded RNA (dsRNA). The box plots show the median and interquartile range, and the bars show the 10th and 90th percentiles. *p<0.05; **p<0.01 vs control untreated cells (Wilcoxon rank sum test). Between-group comparisons were made using the Mann–Whitney U test.
Effects of inhibitors of TLR3, PKR, protein synthesis and IFNα/β on dsRNA-induced TSLP mRNA
To study the mechanisms involved in the induction of TSLP mRNA by dsRNA, several classes of pharmacological inhibitors were tested initially using BECs from healthy subjects. This showed that induction of TSLP mRNA was dose-dependently inhibited by the addition of chloroquine, which inhibits TLR3 by preventing acidification of the endosomes,25 and by a PKR inhibitor (PKRi)26 (figure 4A,B). Similar results were obtained with BECs from subjects with asthma: TSLP mRNA was inhibited by 69.8±5.2% (n=4) with chloroquine (10 μg/ml) and 93.5±5.0% (n=3) with PKRi (1 μM). Neither chloroquine nor PKRi affected cell morphology (see figure E5 in online supplement), and neither inhibitor significantly reduced the induction of IL-8 by combined treatment with TNFα and IL-4 (each at 50 ng/ml) (3.0±1.4-fold induction vs 4.4±2.7-fold induction for chloroquine (10 μg/ml) or 3.3±1.8-fold induction for PKRi (1 μM)). The specificity of the response was further demonstrated using the negative control compound for the PKRi, which had no significant inhibitory effect on TSLP expression. In addition, TSLP gene expression was found to be dependent on de novo protein synthesis as evidenced by inhibition using cycloheximide (figure 4C), but was not dependent on the autocrine or paracrine actions of type I IFNs released in response to exposure to dsRNA, as a neutralising antibody to the type I IFN receptor had no effect on TSLP mRNA expression (figure 4D). Cycloheximide did not inhibit induction of IL-8 by combined treatment with TNFα and IL-4.

Effect of inhibitors on the double-stranded RNA (dsRNA)-induced thymic stromal lymphopoietin (TSLP) response. Bronchial epithelial cells (BECs) were preincubated for 2 h with (A) chloroquine (Chlq) or (B) protein kinase inhibitor (PKRi) before stimulation with dsRNA. (C) Cycloheximide (CHX) was added 1 h before dsRNA and (D) type I interferon (IFN) receptor antibody (R Ab) was added at the same time as dsRNA. Cells were harvested at 3 h and TSLP gene expression was determined by RTqPCR. Data are presented as percentage stimulation in relation to TSLP mRNA expression for cells stimulated with dsRNA. The results are shown as mean±SEM from at least four independent experiments. *p<0.05 vs dsRNA-treated cells (Wilcoxon signed rank test). In (D), data from BECs from healthy subjects and those with asthma are combined owing to the small numbers of BECs tested.
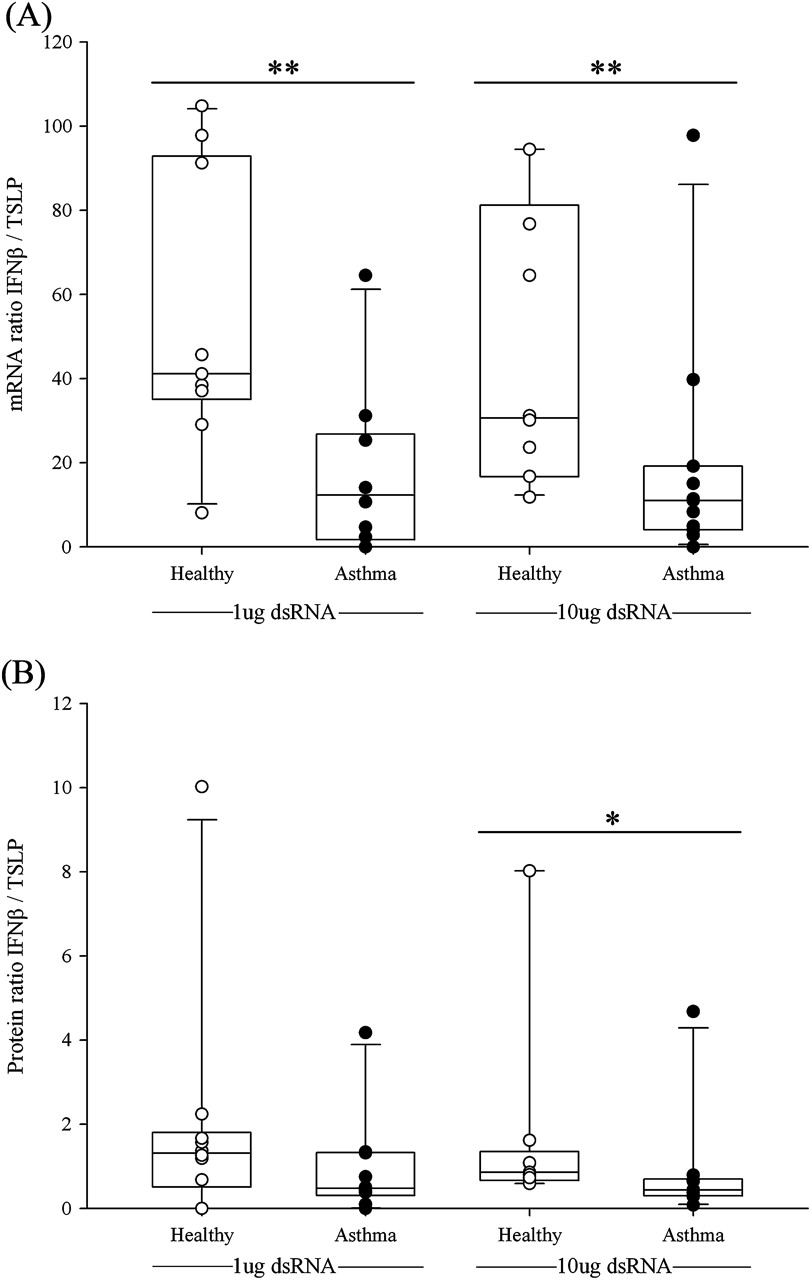
IFNβ/TSLP ratios
As IFNβ can drive a Th1 response27 whereas TSLP favours Th2 responses,15 the reduced generation of IFNβ together with increased levels of TSLP in BECs from subjects with asthma in response to dsRNA made it of interest to calculate the ratio of IFNβ to TSLP on a per subject basis. At 3 h the mRNA ratio was significantly higher in the healthy group at both doses of dsRNA (figure 5A) (healthy vs asthma groups: median values 41 vs 14 (p<0.01) or 31 vs 11 (p<0.01) for 1 and 10 μg/ml dsRNA, respectively). At the protein level the ratios for IFNβ/TSLP were also significantly higher in the healthy group (figure 5B) (healthy vs asthma groups: median values 1.31 vs 0.50 or 0.96 vs 0.43 (p<0.007) for 1 and 10 μg/ml dsRNA, respectively).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of (A) mRNA interferon β/thymic stromal lymphopoietin (IFNβ/TSLP) ratios or (B) protein IFNβ/TSLP ratios in bronchial epithelial cells from healthy subjects (open circles) or subjects with asthma (closed circles) in response to 1 μg/ml or 10 μg/ml double-stranded RNA (dsRNA) calculated on a per subject basis. *p<0.05; **p<0.01 vs control untreated cells (Wilcoxon rank sum test). Between-group comparisons of the ratios were made using the Mann–Whitney U test.
Discussion
Previous studies have shown that exposure to RV infection or dsRNA in vitro induces TSLP production in BECs from healthy subjects,14 18 suggesting that this cytokine may link the innate antiviral response and the type 2 adaptive immune response. As BECs from subjects with asthma have a deficient innate immune response to RV infection,7 we tested the hypothesis that expression of both IFNβ and TSLP is abnormal in asthma-derived BECs following stimulation with dsRNA. This hypothesis is strongly supported by our findings. In primary cultures of BECs obtained from 13 subjects with asthma and 13 healthy controls, dsRNA induced greater expression and release of TSLP in BECs from subjects with asthma than those obtained from healthy individuals. Our studies also showed a fundamental epithelial deficiency in generation of IFNβ in response to a generic TLR3 ligand in BECs from subjects with asthma. Our data therefore support and extend the earlier studies showing deficient IFNβ expression in response to RV infection.7 In contrast with our findings for TSLP and IFNβ, there was no difference between BECs from healthy controls or subjects with asthma with regard to dsRNA-induced epithelial generation of IL-8. This observation agrees with previous studies of other inducers of IL-8 expression including RV,9 as well as house dust mite allergen, TNFα and IL-13.24
TSLP has been advanced as a key new cytokine with significant potential for initiating and aggravating asthma.13 14 17 19 Ying and co-workers13 have observed that airway epithelial TSLP mRNA expression is increased in severe asthma. Although their patients were clinically free from respiratory infection, it cannot be excluded that a viral component could have contributed to the increased epithelial expression of TSLP in that study. The modest TSLP expression at baseline in the epithelium of subjects with asthma in our study is consistent with the findings of Ying et al13 who observed relatively little epithelial expression of TSLP in the bronchi of subjects with asthma with approximately the same degree of bronchial obstruction (percentage predicted forced expiratory volume in 1 s) as that recorded in the subjects with asthma in our study. Our TSLP data further agree well with previous findings using commercially sourced epithelial cells. Thus, Kato et al18 reported viral stimulus-induced expression and release of TSLP from normal BECs, and Allakhverdi et al14 reported dsRNA-induced release of TSLP from small airway epithelial cells. However, Lee and Ziegler,19 working with normal human BECs (sourced from Clonetics) observed no effect of 5 μg/ml dsRNA on TSLP mRNA expression 2 h after stimulation. This latter negative finding disagrees with the present study as well as with the findings of Kato et al,18 both of which studies demonstrated peak increases in TSLP mRNA 3 h after dsRNA stimulation. On the other hand, Kato et al18 did not observe any increase in TSLP mRNA even at 100 ng/ml TNFα whereas Lee and Ziegler19 demonstrated increased expression and production of TSLP in response to 10 ng/ml TNFα.
Previous investigations of the mechanisms leading to induction of TSLP mRNA expression by dsRNA have shown the involvement of TLR3, nuclear factor kappa B (NFκB) and interferon regulatory factor 3 (IRF3) in this response.18 In agreement with these previous findings, the present study demonstrated inhibition of TSLP expression with chloroquine, suggesting the involvement of TLR3, while the observed inhibition of PKR, whose substrates include IκB, would limit activation of NFκB. Although it is possible that these inhibitors had non-specific toxic effects, this is unlikely as the BECs showed no morphological evidence of toxicity and there was no effect of the inhibitors on TNFα/IL-4-induced IL-8 expression. Our data are consistent with the report by Gern et al20 which showed that the PKR inhibitor 2-aminopurine inhibited dsRNA-induced increases in other NFκB-regulated cytokines such as CCL5 and IL-8 in human BECs. Although Kato et al18 did not observe inhibition of dsRNA-induced TSLP expression in cells treated with siRNA against PKR, this may be due to insufficient knockdown of the PKR protein. As activation of PKR by dsRNA induces phosphorylation of the translation initiation factor EIF2α28 leading to inhibition of the initiation of protein synthesis, our findings that TSLP expression is dependent on de novo protein synthesis also provides indirect support for the involvement of PKR. Since TLR3, IRF3 and NFκB are required for induction of both TSLP and IFNβ expression, the preferential induction of TSLP versus IFNβ in BECs from subjects with asthma is a conundrum. It seems unlikely that TSLP production is driven by autocrine release of IFNβ as neutralisation of the type I IFN receptor did not affect TSLP expression. In view of the requirement for de novo protein synthesis, it is possible that specific post-transcriptional events are key to understanding the differential regulation of TSLP and IFNβ expression. Further studies will be required to elucidate these mechanisms and to help explain the abnormal overproduction of TSLP and the underproduction of IFNβ in the epithelium of subjects with asthma.
The inability of BECs from subjects with asthma to mount a normal antiviral response to RV16 infection has been reported for the induction of both IFNβ7 29 and IFNλ,8 although two more recent studies failed to observe this difference.30 31 In the study by Lopez-Souza et al,30 the absence of a disease-related difference may be explained by the fact that the BECs were differentiated at an air-liquid interface and were substantially more resistant to infection. Although Bochkov et al31 used undifferentiated BEC cultures similar to those used in the current study and the earlier studies of RV infection,7–9 they infected the cells using a minor group RV using a large innoculum (10 plaque forming units/cell). This led to substantial cytopathic cell death effects by 18 h of infection, and there was little evidence of an antiviral response which suggests that the BECs had died rapidly before an innate immune response could be mounted. In contrast, in the study by Wark et al,7 little cell death was evident, especially in the healthy control BEC cultures. A differential effect of viral titre on the antiviral response has been observed using West Nile virus, where the cells died rapidly by necrosis (evident at 8 h) when infected with a multiplicity of infection ≥10 whereas, at lower infectious doses, death was by apoptosis and occurred much more slowly (32 h after infection).32 In our current studies using synthetic dsRNA to activate TLR3, the innate antiviral response was not confounded by any significant cell death and we found lower IFNβ mRNA and protein expression in BECs from subjects with asthma. Furthermore, in the studies by Bochkov et al31 the BECs were all from subjects with mild asthma whereas the initial study by Wark et al7 reported that BECs from corticosteroid-treated subjects with asthma were more permissive for RV replication than those from subjects with mild asthma. In the current study, seven of the 13 subjects with asthma were treated with corticosteroids and subgroup analysis suggested a trend for a lower dsRNA-induced IFNβ response in this group compared with the six donors whose asthma did not require corticosteroid treatment.
Recent studies suggest that IFNβ production by BECs is a key driver for normal DC maturation towards one capable of eliciting a protective Th1 immune response.33 In contrast, TSLP-activated DCs do not produce detectable proinflammatory cytokines but do produce high levels of TARC (CCL17) and MDC (CCL22) chemokines which preferentially attract CCR4-expressing Th2 lymphocytes.10 These DCs also induce marked proliferation and expansion of allogeneic-naїve CD4+ T cells which produce large amounts of IL-13, IL-5 and TNFα but only small amounts of IL-10 and IFNγ.10 16 34 Stimulation of DC TLR3 could generate a Th1 polarising signal (IL-12) but, in previous studies, dsRNA did not prevent TSLP-activated DCs from priming naïve CD4+ T cells to expand into inflammatory Th2 cells.34 Our observation that dsRNA-treated BECs from subjects with asthma show a deficiency in mobilising an antiviral host response (IFNβ) in combination with overproduction of a potentially disease-initiating cytokine (TSLP)10 15 16 underscores the potential role of an abnormal epithelium in response to early life virus infections leading to deviation of T cell maturation along a proallergic Th2 pathway.
Although increased epithelial TSLP expression has been observed in severe asthma,13 17 it is not known to what extent the present observations translate into in vivo airway epithelium in health and disease. One possible criticism is that the cells used in the present study were undifferentiated; however, this may reflect damaged and repairing epithelium which is readily apparent in asthma.35 Further studies of differentiated epithelial cell models may provide additional insight into the effect of differentiation status on TSLP expression and the persistence of disease-related differences in this context. In addition, although the observations in the present study were made using dsRNA as a mimic of danger responses of viral RNA, it will be important to extend these studies towards virus infection models and the consequence of epithelial infection on DC activation and T cell polarisation.
In conclusion, the use of dsRNA poly I:C as a stimulus has allowed us to demonstrate the reduced innate immune response of BECs from subjects with asthma independent of the severity of infection. As such, it provides a useful model for further exploratory studies. Our study shows a dual abnormality of BECs from subjects with asthma in response to dsRNA involving reduced generation of a major antiviral interferon (IFNβ) and overproduction of a potentially disease-initiating cytokine (TSLP). The relationship between these two proteins in the epithelium of patients with asthma translates as an unfavourable ratio significantly different from that in the epithelium of those without asthma. These data support the possibility that intervention with TSLP inhibitory treatments and/or supplementary IFNβ administration to the bronchial mucosa may reduce the occurrence and severity of RV-induced exacerbations of asthma.
References
Supplementary materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Funding Medical Research Council (MRC) UK, Swedish Medical Research Council, Swedish Heart-Lung Foundation and VINNOVA.
Competing interests None.
Ethics approval Written informed consent was obtained from all volunteers before entering the study and all procedures were approved by the Southampton and South West Hampshire Research Ethics Committee (REC number 05/Q1702/165).
Provenance and peer review Not commissioned; externally peer reviewed.