Article Text

Abstract
Background and aim Alveolar fluid clearance is impaired by inducible nitric oxide synthase (iNOS)/nitric oxide (NO)-dependent mechanisms in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS). The activation of the stress protein response (SPR) in alveolar macrophages on iNOS-dependent NO production in response to interferon γ (IFNγ), a major cytokine present in the airspace of patients with ALI, was investigated.
Methods The SPR was activated in murine and primary human alveolar macrophages prior to analysis of signal transducer and activator of transcription factor 1 (STAT1) activation, iNOS mRNA and protein synthesis, and NO production.
Results SPR activation resulted in inhibition of IFNγ-mediated NO production (p=0.001) with >95% detergent insolubilisation of the STAT1 protein. Its subsequent proteasomal degradation was partially reversed with pretreatment of cells with the chemical chaperone glycerol. This early effect of the SPR was caused by the complete disruption of heat shock protein 90 (Hsp90)–STAT1 binding, as shown by immunoprecipitation. Recovery of STAT1 activation and recovery of iNOS synthesis occurred within 12 h after SPR activation (p=0.02). NO production (as compared with non-SPR controls) did not occur until 48 h later (p=0.02). SPR-induced Hsp70 (Hsp70i) expression caused a late inhibition of NO production (p=0.02). Inhibiting >50% Hsp70i expression recovered NO production to control levels whereas overexpressing Hsp70i in the absence of the SPR inhibited NO production (p=0.02).
Conclusion Early inhibition of STAT1 following its dissociation from Hsp90, and later inhibition of iNOS activity by Hsp70i, represent novel mechanisms by which SPR activation modulates the IFNγ signalling in alveolar macrophages. These results highlight a potential clinical application for Hsp90 inhibitors in modulating NO signalling during the early phase of acute lung injury.
- Chaperones
- heat shock
- lung
- innate immunity
- macrophage biology
- macrophages
- signal transduction
- STAT1–Hsp90 interaction
Statistics from Altmetric.com
- Chaperones
- heat shock
- lung
- innate immunity
- macrophage biology
- macrophages
- signal transduction
- STAT1–Hsp90 interaction
Introduction
Acute lung injury (ALI) is a devastating clinical syndrome manifested by an inflammatory response leading to respiratory failure with an overall mortality rate of 30–40%.1 Clinical studies have shown that the vectorial fluid transport across the alveolar epithelium critical for maintaining fluid balance in the airspace is impaired in most patients with ALI.1 There is a direct correlation between the impairment of fluid removal from the airspace and mortality in patients with ALI.2 Recent experimental evidence indicates that alveolar fluid clearance (AFC) is impaired by inducible nitric oxide synthase (iNOS)/nitric oxide (NO)-dependent mechanisms.3 Under these circumstances, NO released by alveolar macrophages and epithelial cells modifies ENaC (epthelial sodium channel), CFTR (cystic fibrosis transmembrane conductance regulator) and other ion channels involved in alveolar epithelial ion transport, rendering these ion channels non-functional.4 5 iNOS-dependent NO production in the airspace of patients with ALI depends on the release of several inflammatory mediators including interferon γ (IFNγ) that has been found in high levels in the bronchoalveolar lavage (BAL) of these patients and animal models of ALI.6 7 IFNγ activates signal transducer and activator of transcription factor 1 (STAT1), which activates transcription of various genes including that encoding iNOS.8
The heat shock or stress protein response (SPR), a highly conserved cellular defence mechanism, is part of the early lung innate immune response to injury.9 The heat shock response is characterised by an early phase defined by the inhibition of proinflammatory cell signalling pathways within minutes after onset of the SPR and a delayed phase in which the development of tolerance to inflammatory stimuli takes several hours and requires de novo protein synthesis including the expression of inducible heat shock proteins Hsp70 and Hsp27.10 We recently reported that SPR activation does occur in patients with ALI and correlates with the preservation of AFC.9 Furthermore, we have previously shown that SPR activation restored AFC in animal models of haemorrhagic shock and ischaemia–reperfusion injury in part by reducing iNOS synthesis.11 12 In some of these studies, the inhibitory effect of SPR activation could be reproduced by the adenoviral gene transfer of Hsp70 into the distal airspaces of the lung, suggesting that Hsp70 may participate in the anti-inflammatory effect induced by SPR activation.13 Despite these results, the mechanisms by which SPR activation affects the iNOS-dependent NO release are still not completely understood. Thus, the objective of this study was to determine the mechanisms by which SPR activation regulates the STAT1 signalling pathway and the subsequent production of NO by murine and human alveolar macrophages after exposure to IFNγ.
Materials and methods
For full details, see the supplementary methods online.
Cells
A murine alveolar macrophage cell line, MH-S, and primary human alveolar macrophages were used in these studies.
SPR activation using heat (heat stress)
MH-S cells were incubated for 30 min and human macrophages for 60 min at 43°C and allowed to recover at 37°C for the times indicated in the text.
Western blotting
The cells were washed three times with phosphate-buffered saline (PBS) on ice and lysed in 1× Laemmli sample buffer (LSB). The protein concentrations were determined using the BioRAD protein assay (Bio-Rad, Hercules, California, USA). Equal amounts of protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to nitrocellulose. Antibody dilutions used to detect protein expression are indicated in the figure legends.
Measurement of extracellular NO
Cells were stimulated with IFNγ for 24 h. The presence of nitrite in the extracellular medium was determined using the Griess method.
Measurement of iNOS or inducible Hsp70 (Hsp70i) mRNA
Real-time reverse transcription–PCR (RT–PCR) primers and probes were designed using Primer Express software (PE-Applied Biosystems (PE-ABI), Warrington, UK). The probes were labelled with a fluorophore reporter dye (6-carboxy-fluorescein) at the 5′ end and a quencher dye (BHQ (black hole quencher); Biosearch Technologies, Novato, California, USA) at the 3′ end. Quantitative real-time RT–PCR was performed as previously published.14 The number of cycles to threshold (CT) of fluorescence detection was normalised to the CT of glyceraldehyde phosphate dehydrogenase (GAPDH) for each sample tested. Mouse RT–PCR iNOS primers were; TAGF, GAGCATCCCAAGTACGAGTGGT; TAGR, GGCCACGGCAGGCAG; and TAGP, CCAGGAGCTCGGGTTGAAGTGGTATG. Hsp70i primers were: TAGF, CTGTAGGAAGGATTTGTACACTTTAAACTC; TAGR, TTAAGGGTCAGCTCCTGAAGGT; and TAGP, TCTGAGTCCCACACTCTCACCACCCA. Human RT–PCR iNOS primers were: TAGF, AGCGGCTCCATGACTCTCA; TAGR, TGCACCCAAACACCAAGGT and TAGP, AGAGGGCTCAAAGGAGGCCGCAT.
EMSA
Nuclear protein isolation and EMSA were performed as described previously.15 EMSA was performed using a double-stranded oligonucleotide purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA) that contains a consensus binding site for STAT1 (sc-2537 GAS/ISRE (γ-activation sequence/interferon-simulating response element oligonucleotide))16 17 and was end labelled with [γ-32P]ATP.
Isolation of the membrane-enriched fraction
Cells were scraped into a hypotonic buffer (10 mM HEPES, 10 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, protease inhibitors and phosphatase inhibitors) and processed as previously described.18
Immunoprecipitations
Cells were lysed in 50 mM Tris, 150 mM NaCl, 1% Triton, 40 mM sodium molybdate and protease inhibitors. An antibody to STAT1 was added to immunoprecipitate STAT1–Hsp90 complexes at 4°C.
Small interfering RNA (siRNA) transfection
siRNA to Hsp70i was transfected into cells using X-tremeGene according to the manufacturers protocol using a ratio of 10:2 of transfection reagent in microlitres to micrograms of siRNA.
Adenovirus infection
Adenovirus infection of macrophages was performed as previously described.19
Statistics
Data analysis was performed by the investigators. All data were tested for normality and, due to non-uniform distribution, the data were subsequently analysed with non-parametric tests. Data were calculated as medians±IQRs. The data are presented in the figures as box plots with IQRs and lower and upper ranges. The Kruskal–Wallis test followed by the Dunn test were used to compare three or more experimental groups. The Mann–Whitney test was used to compare two experimental conditions. A p value of <0.05 was considered statistically significant.
Results
SPR activation inhibits IFNγ-mediated NO production in murine macrophages
Alveolar macrophages exposed to 5 min increments of heat stress at 43°C and recovered at 37°C for 1 h before IFNγ stimulation for 24 h released decreasing amounts of NO (nitrite) measured in the extracellular medium (5, 10, 15, 20 and 25 min (p=0.01) compared with no heat shock and IFNγ stimulation) (figure 1A). After 30 min of heat stress and 1 h recovery, NO production was completely inhibited (p=0.01) compared with no heat shock. This decrease in NO production was not due to cell death as determined by Alamar blue on the same cells that were heat stressed and treated with IFNγ (p=0.38) (figure 1B). Further experiments demonstrated that after a 30 min SPR activation, IFNγ stimulation did not induce iNOS mRNA synthesis (p=0.04) (figure 1C) or protein expression (figure 1D).

Stress protein response (SPR) activation inhibits interferon γ (IFNγ)-mediated nitric oxide (NO) production, inducible nitric oxide synthase (iNOS) mRNA and protein in alveolar macrophages. (A) MH-S cells were incubated at 43°C for increasing times and allowed to recover at 37°C for 1 h prior to IFNγ stimulation (10 ng/ml) for 24 h. Extracellular medium was assayed for nitrite using the Griess reagent. *p=0.01 from control cells; **p=0.01 from cells stimulated with IFNγ alone. (B) Cell viability of MH-S cells was determined using the Alamar blue assay (p=0.38). For both A and B, results are the median±IQRs of four separate experiments with each condition carried out in triplicate. (C) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 2 h) and analysis for iNOS mRNA by real-time reverse transcription–PCR (RT–PCR) normalised with glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA levels. Results are the median±IQRs of three experiments done in triplicate; *p=0.03 from control cells; **p=0.04 from cells treated with IFNγ alone. (D) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 7 h) and analysed for iNOS protein by western blot; one representative experiment is shown; three additional experiments gave comparable results. HS, heat shock; Rec, recovery.
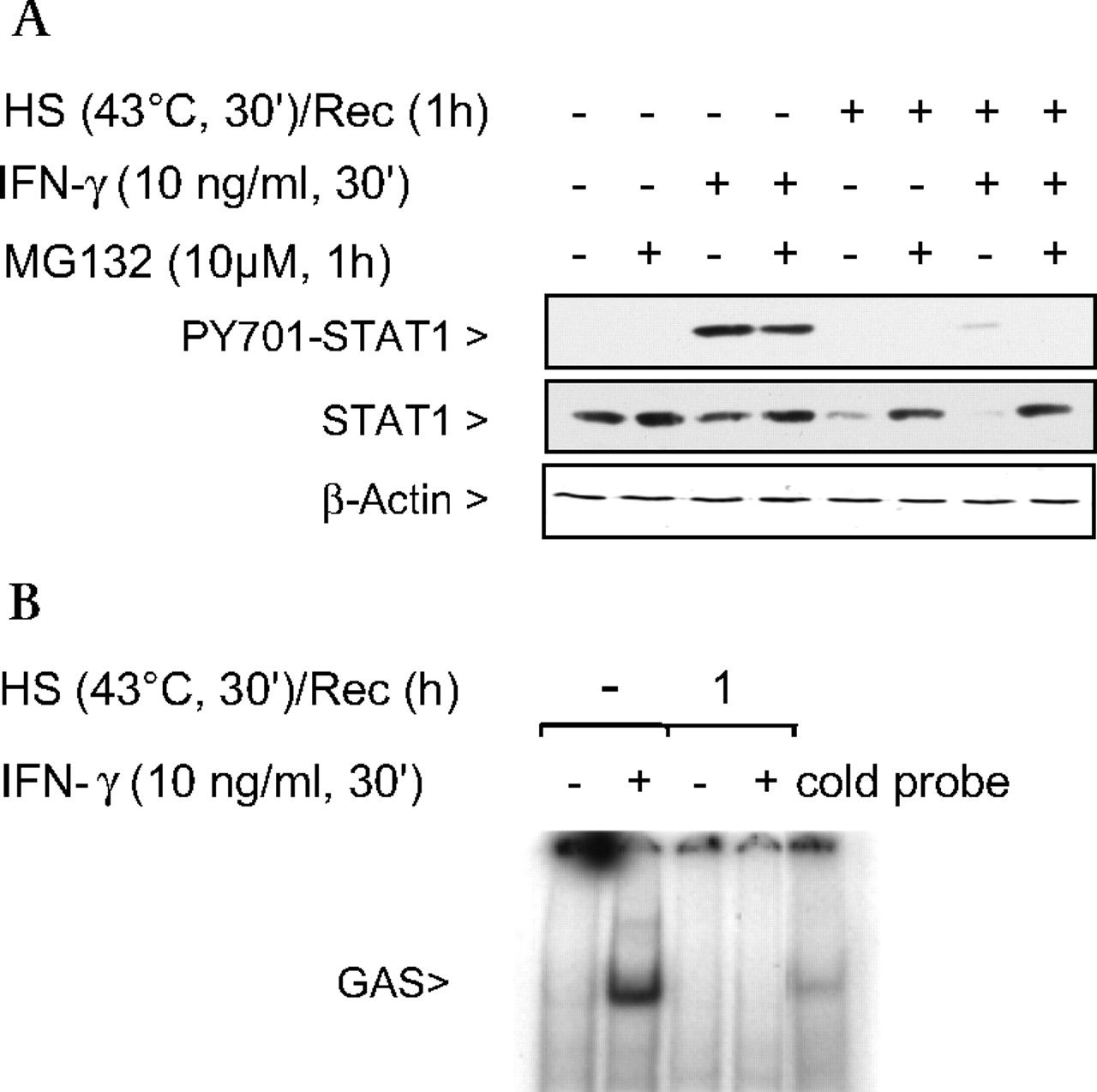
STAT1 stimulated by IFNγ is an essential activator of iNOS transcription.20 Activation of STAT1 results in some proteosomal degradation of the protein; however, STAT1 phosphorylation and signalling were not affected (figure 2A, lane 3). In contrast, when cells were heat stressed, there was >95% proteosomal degradation (figure 2A, lanes 5 and 7) of STAT1, and STAT1 phosphorylation on Tyr701 was inhibited compared with no heat stress and IFNγ stimulation (figure 2A). This difference can be explained by the fact that IFNγ-mediated STAT1 activation results in normal turnover of the protein whereas SPR activation results in denaturation and insolubility of STAT1, rendering STAT1 unavailable for activation by IFNγ (Supplementary figure S1A). STAT1 insolubility due to protein denaturation can be prevented with pretreatment of the cells with glycerol, a chemical chaperone that reduces heat-induced protein denaturation (Supplementary figure S1B). In addition, SPR activation prevented STAT1 translocation to the nucleus. As shown in figure 2B, in the absence of heat stress, STAT1 protein extracted from the nucleus after IFNγ stimulation bound to the STAT1 GAS/ISRE oligonucleotide whereas after heat stress, a 1 h recovery and IFNγ stimulation, no binding was observed.

Stress protein response (SPR) activation prevents interferon γ (IFNγ)-mediated activation of signal transducer and activator of transcription factor 1 (STAT1). (A) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 30 min) and then analysed for STAT1 and phosphorylated STAT1 by western blot. Some cells were pretreated with a proteasome inhibitor MG132 (10 μM) or the vehicle 1 h prior to heat shock and exposure to IFNγ. Representative blot of two experiments. (B) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 30 min). EMSA analysis was used to examine STAT1 activity in IFNγ-treated cells. Nuclear extracts were prepared (see the Materials and methods section) and 5 μg of nuclear protein were incubated with a 32P-labelled DNA containing the STAT1 binding consensus sequence (γ-activation sequence (GAS)). In some reactions, a 100-fold excess of the unlabelled oligonucleotide was added (indicated as cold probe in the figure). The experiment was performed five times. One representative experiment is shown. HS, heat shock; PY701, STAT1 phosphorylated on Tyr701; Rec, recovery.
SPR activation prevents IFNγ-mediated activation of STAT1, iNOS mRNA synthesis and NO production in primary human alveolar macrophages
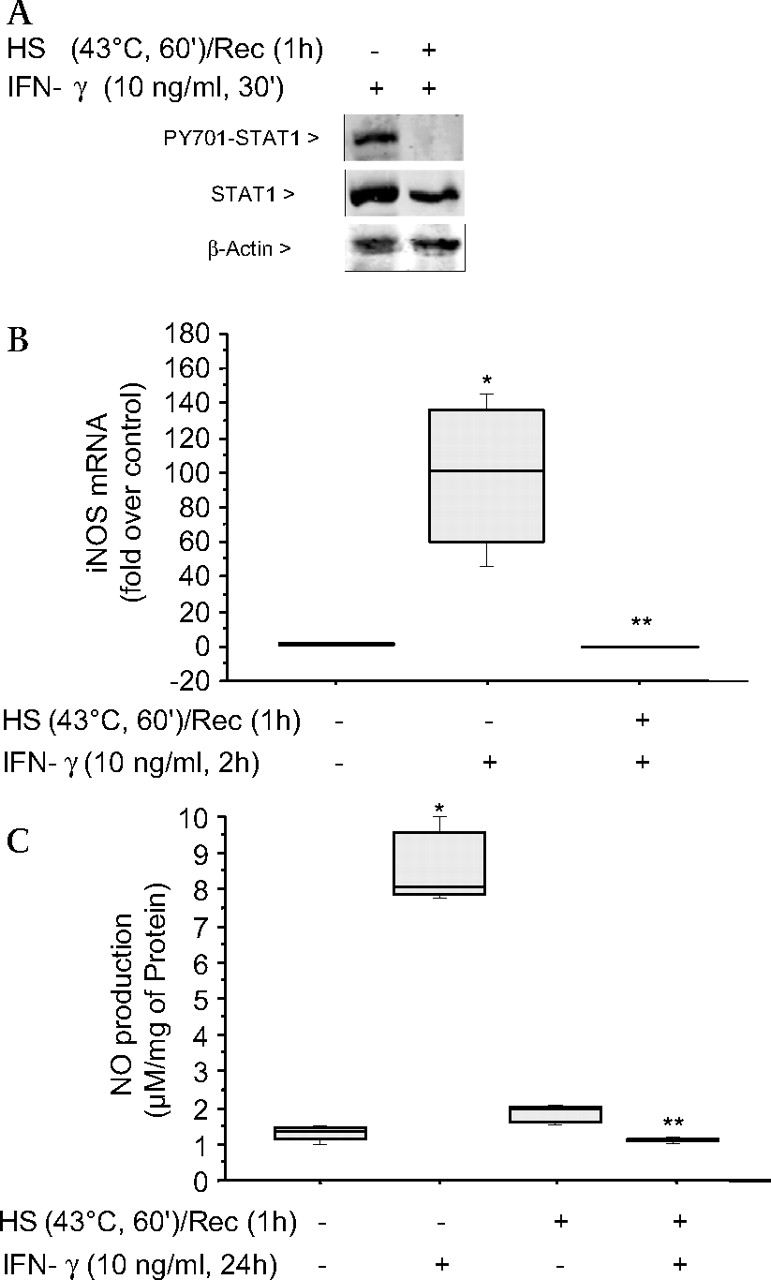
Further studies demonstrated that SPR activation had the same effect on the IFNγ signalling pathway in human alveolar macrophages as observed in the MH-S cell line. Human alveolar macrophages were heat stressed for 60 min prior to IFNγ stimulation for 30 min. STAT1 phosphorylation was inhibited (figure 3A), with subsequent inhibition of IFNγ-mediated iNOS mRNA synthesis (p=0.04) (figure 3B) and NO production (p=0.04) (figure 3C).

The stress protein response (SPR) prevents signal transducer and activator of transcription factor 1 (STAT1) phosphorylation, inducible nitric oxide synthase (iNOS) mRNA synthesis and nitric oxide (NO) production in primary human alveolar macrophages. (A) Human macrophages were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to interferon γ (IFNγ) stimulation (10 ng/ml for 30 min) and then analysed for STAT1 and phosphorylated STAT1 by western blot. (B) Human macrophages were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 2 h) and analysed for iNOS mRNA by TAQman. (C) Human macrophages were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 24 h) and analysed for NO production. For B and C, the results are the median±IQRs of two separate experiments with each condition carried out in triplicate; *p=0.04 from control cells; **p=0.04 from cells treated with IFNγ alone. HS, heat shock; PY701, STAT1 phosphorylated on Tyr701; Rec, recovery.
STAT1 function requires Hsp90
Hsp90 is known to bind various proteins to maintain protein conformation necessary for function. We tested whether STAT1 was an Hsp90 client protein. Hsp90 co-immunoprecipitated with STAT1 under non-stimulated conditions in MH-S cells, but not after SPR activation with heat (figure 4A). Furthermore, both STAT1 and Hsp90 were recruited to the membrane-enriched fraction 10–15 min after exposure to IFNγ and this was associated with an increase in the level of STAT1 phosphorylated on Tyr701 (figure 4B). Treatment of macrophages for 1 h with 17-AAG (17-allylamino-17-demethoxygeldanamycin), an inhibitor of Hsp90 binding, and subsequent IFNγ stimulation resulted in a decrease in STAT1 phosphorylation on Tyr701 using 5 μg/ml of 17-AAG (5 μg/ml=IC50), and a >95% decrease in phosphorylation using 50 μg/ml of 17-AAG (p<0.01) (figure 4C). This loss of activated STAT1 by 17-AAG correlated with a 60% (p=0.04) and 95% decrease (p=0.04) of iNOS gene transcription, respectively (figure 4D).

Signal transducer and activator of transcription factor 1 (STAT1) forms a complex with heat shock protein 90 (Hsp90) and is disrupted by stress protein response (SPR) activation in alveolar macrophages. (A) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 1 h. The cells were then lysed in a buffer containing 0.1% NP-40. Following centrifugation, the resultant supernatants were used for immunoprecipitation (IP) reactions using an antibody specific for STAT1. The resultant immunoprecipitates were examined via western blotting for Hsp90 and STAT1. (B) MH-S cells were stimulated with interferon γ (IFNγ) (10 ng/ml) for the indicated times. Plasma membranes were isolated using Dounce homogenisation and differential centrifugation. The presence of total STAT1, phosphorylated STAT1 and Hsp90 was determined in the cell membrane fraction by western blot. (C) MH-S cells were pretreated with 17-allylamino-17-demethoxygeldanamycin (17-AAG), an Hsp90 inhibitor, (5 or 50 μg/ml), 1 h prior to IFNγ stimulation (10 ng/ml for 30 min) and then analysed for STAT1 and phosphorylated STAT1 by western blot. Each set of experiments has been performed with the same final concentration of dimethylsulfoxide (DMSO; solvent for 17-AAG). (D) MH-S cells were pretreated with 17-AAG (5 and 50 μg/ml) 1 h prior to IFNγ stimulation (10 ng/ml for 2 h) and analysed for inducible nitric oxide synthase (iNOS) mRNA by real-time reverse transcription–PCR (RT–PCR) normalised with glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA levels. Each set of experiments has been performed with the same final concentration of DMSO (solvent for 17-AAG). Results are the median±IQRs of three experiments done in triplicate; *p=0.04 from control cells; **p=0.04 from cells treated with IFNγ alone. HS, heat shock; IB, immunoblot; PY701, STAT1 phosphorylated on Tyr701; Rec, recovery.
Recovery of NO production and IFNγ-stimulated STAT1 signalling post-SPR activation
NO production from heat-stressed cells that was not statitistically different from NO production from control cells was only observed after 48 (p=0.14) and 72 h (p=0.38) following heat stress (figure 5).
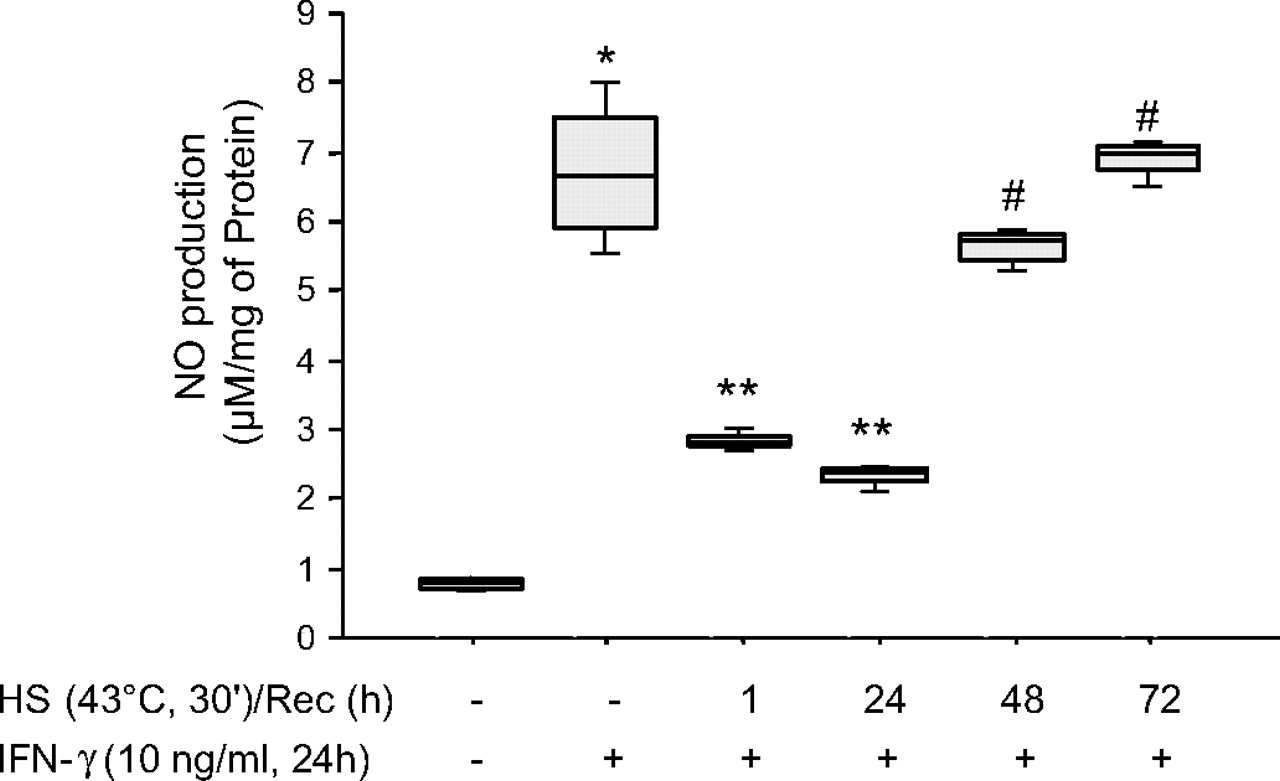
Inducible nitric oxide synthase (iNOS) function recovers 48 h after stress protein response (SPR) activation. MH-S cells were incubated at 43°C for 30 min, then allowed to recover at 37°C for the indicated times prior to interferon γ (IFNγ) stimulation (10 ng/ml) for 24 h. Extracellular medium was assayed for nitrite using the Griess reagent. Results are the median±IQRs of four separate experiments with each condition carried out in triplicate; *p=0.02 from control cells; **p=0.02 from cells treated with IFNγ alone; #p=0.02 from cells heat stressed/recovered for 1 h and treated with IFNγ. HS, heat shock; Rec, recovery.
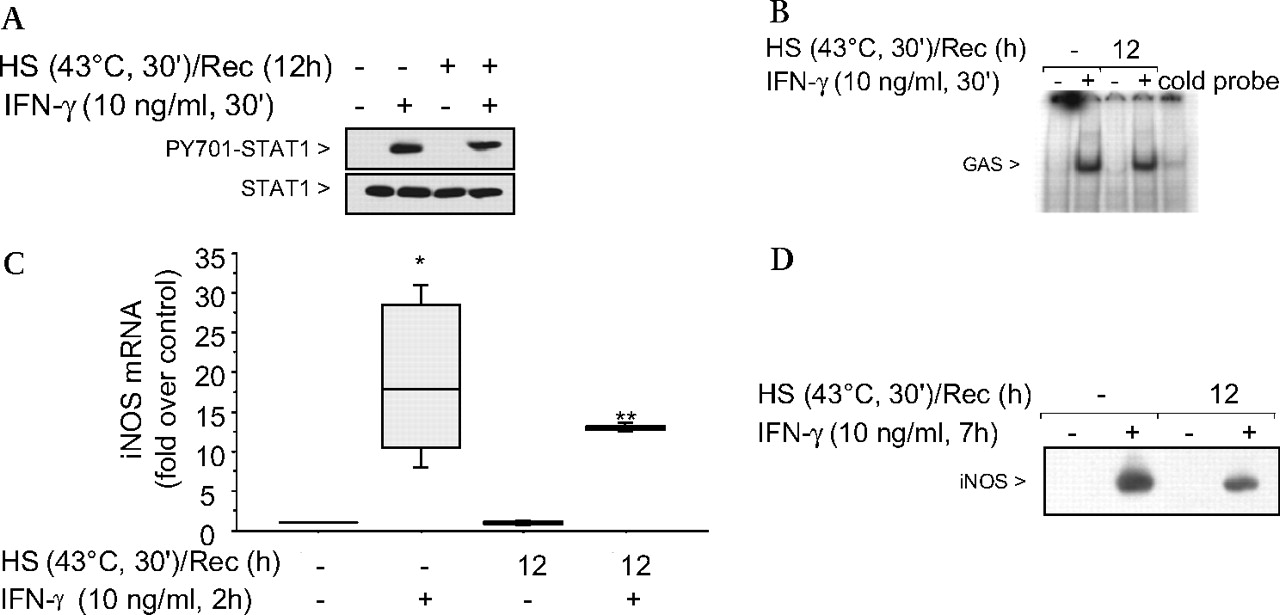
We determined whether the slow recovery of the IFNγ-mediated NO production after SPR activation was caused by a prolonged inhibition of the STAT1 pathway. Figure 6A shows that with a 12 h recovery postheat stress, the STAT1 protein was phosphorylated to the same level as non-heat-stressed cells. Since the STAT1 protein was phosphorylated, the presence of nuclear STAT1 protein and its DNA binding function after a 12 h recovery from heat stress was tested. As shown in figure 6B, in the absence of heat stress, STAT1 protein extracted from the nucleus after IFNγ stimulation bound to the STAT1 GAS/ISRE oligonucleotide. Likewise, after heat stress, 12 h recovery and IFNγ stimulation, STAT1 binding comparable with levels from control cells was observed. Further analysis confirmed that the IFNγ-dependent activation of the STAT1 signalling pathway fully recovered after 12 h recovery from heat stress as iNOS mRNA levels similar to levels seen in non-heat-stressed IFNγ stimulated cells were detected (p=0.44) (figure 6C) as well as the iNOS protein (figure 6D). These results show that after a 12 h recovery from heat stress, alveolar macrophages stimulated with IFNγ are able to activate the STAT1 pathway, resulting in synthesis of both iNOS mRNA and protein. In contrast, the release of NO from these cells after SPR activation and subsequent IFNγ stimulation did not follow the same temporal recovery. Indeed, there was an inhibition of NO production 12 h after SPR activation even though the iNOS mRNA and protein were synthesised. Thus, the SPR regulates iNOS-dependent NO release by mechanisms that are independent of iNOS gene and protein expression.

The signal transducer and activator of transcription factor 1 (STAT1) signalling pathway is functional after 12 h recovery postheat stress induction and subsequent interferon γ (IFNγ) stimulation. (A) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 12 h prior to IFNγ stimulation (10 ng/ml for 30 min) and then analysed for STAT1 and phosphorylated STAT1 by western blot. (B) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 12 h prior to IFNγ stimulation (10 ng/ml for 30 min). EMSA analysis was used to examine STAT1 activity in IFNγ-treated cells. Nuclear extracts were prepared (see the Materials and methods section) and 5 μg of nuclear protein were incubated with a 32P-labelled DNA containing the STAT1 binding consensus sequence (γ-activation sequence (GAS)). In some reactions, a 100-fold excess of the unlabelled oligonucleotide was added (indicated as cold probe in the figure). The experiment was performed five times. One representative experiment is shown. (C) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 12 h prior to IFNγ stimulation (10 ng/ml for 2 h) and analysis for inducible nitric oxide synthase (iNOS) mRNA by real-time reverse transcription–PCR (RT–PCR) normalised with glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA levels. Results are the median±IQRs of three experiments done in triplicate; *p=0.02 from control cells; **p=0.02 from heat-stressed cells. (D) MH-S cells were either untreated or were incubated at 43°C and allowed to recover for 1 h prior to IFNγ stimulation (10 ng/ml for 7 h) and analysis for iNOS protein by western blot; one representative experiment is shown; three additional experiments gave comparable results. HS, heat shock; PY701, STAT1 phosphorylated on Tyr701; Rec, recovery.
Since the inducible form of Hsp70 is expressed at high levels after heat stress, we hypothesised that in the late phase of SPR activation, Hsp70i expression transiently inhibits iNOS function. As shown in figure 7A, Hsp70i expression was expressed at high levels after 1 h recovery and these levels significantly decreased by 25% after 24 h recovery (p=0.02) and by >50% after 48 h recovery (p=0.02). The protein was not detected after 72 h recovery. To explore further the importance of SPR-induced Hsp70 expression on iNOS function, Hsp70 expression was inhibited using a specific siRNA. Alveolar macrophages were transfected with an siRNA control or a specific siRNA to Hsp70 after 30 min of heat stress. The cells were allowed to recover for 24 h and then stimulated with IFNγ. As seen in figure 7B, siRNA transfection after heat stress resulted in a 65–70% reduction in Hsp70 expression (p=0.02). This reduction in Hsp70i expression was sufficient to allow NO release by alveolar macrophages stimulated with IFNγ (p=0.02 from cells treated with a control siRNA) (figure 7C). Control and Hsp70i siRNA transfections had no effect on NO production (data not shown). Lastly, we determined whether overexpression of the inducible Hsp70i protein would prevent the release of NO from non-heat-stressed alveolar macrophages when stimulated with IFNγ. Infection of MH-S cells with an adenovirus encoding Hsp70i, but not with a control adenovirus, significantly increased the expression of both Hsp70i mRNA (p=0.03) and protein (figure 7D,E) and decreased the release of NO from these cells after stimulation with IFNγ (p=0.02) (figure 7F).

Inducible heat shock protein 70 (Hsp70i) inhibits inducible nitric oxide synthase (iNOS) function due to activation of the stress protein response (SPR) by heat. (A) MH-S cells were incubated at 43°C for 30 min, then allowed to recover at 37°C for the indicated times prior to interferon γ (IFNγ) stimulation (10 ng/ml) for 24 h. Cells were analysed for Hsp70i by western blot. Expression levels of Hsp70i at the 30 min heat shock and 1 h recovery is set at 100% and all other Hsp70i expression levels are a percentage of that expression level. Results are the median±IQRs of three experiments; *p=0.02 from cells that were subjected to 30 min heat shock and 1 h recovery. (B) MH-S cells were either transfected with a control small interfering RNA (siRNA) or a specific siRNA to Hsp70 24 h before heat stress (before) or were heat shocked, allowed to recover at 37°C for 1 h and then transfected with a control or a specific siRNA to Hsp70 (after). At 24 h postheat stress, cells were lysed and the expression of Hsp70 protein determined by western blot. Densitometry analysis results are the median±IQRs of three experiments; *p=0.02 from cells treated with control siRNA. The expression levels of Hsp70i at the 30 min heat shock and 1 h recovery is set at 100% and all other Hsp70i expression levels are a percentage of that expression level. (C) MH-S cells were incubated at 43°C for 30 min, allowed to recover at 37°C for 1 h and then transfected with a control or a specific siRNA to Hsp70. At 24 h post-transfection, cells were exposed to IFNγ (10 ng/ml) for 24 h. Extracellular medium was assayed for nitrite using the Griess reagent. Results are the median±IQRs of three separate experiments with each condition carried out in triplicate; *p=0.02 from control cells; ** p=0.02 from heat stressed exposed to control siRNA and exposed to IFNγ. (D) Cells were infected with a recombinant adenovirus (Ad) expressing Hsp70i. At 48 h postinfection, cells were lysed and the expression of Hsp70i mRNA determined by real-time reverse transcription–PCR (RT–PCR) normalised with glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA levels. Results are the median±IQRs of three experiments done in triplicate; *p=0.03 from cells treated with control Ad. (E) Cells were infected with a recombinant Ad expressing Hsp70i. At 48 h postinfection, cells were lysed and the expression of Hsp70i protein determined by western blot. (F) Cells were infected as described above. Cells were exposed to IFNγ (10 ng/ml) 24 h postinfection for 24 h. At 48 h postinfection, the extracellular medium was assayed for nitrite using the Griess reagent. Results are reported as nitric oxide (NO) production of infected cells. Results are the median±IQRs of three separate experiments with each condition carried out in triplicate; *p=0.02 from control cells; **p=0.02 from control Ad-infected cells exposed to IFNγ.
Discussion
In this study, we show for the first time that activation of the SPR inhibits IFNγ-mediated activation of the STAT1/iNOS signalling pathway and NO production in murine and human alveolar macrophages. We found that (1) STAT1 and Hsp90 form a complex in the unstimulated cell while IFNγ stimulation led to recruitment of both STAT1 and Hsp90 to the plasma membrane; (2) the disruption of STAT1–Hsp90 binding inhibits IFNγ activation of STAT1 and results in the insolubility and subsequent degradation of STAT1 via the proteosome; (3) the late inhibition of iNOS function after heat stress is due to the expression of inducible Hsp70 and not additional biochemical changes associated with heat stress10; and lastly (4) a non-lethal heat stress inhibits IFNγ-mediated NO production without directing the alveolar macrophages into the apoptotic pathway.
What are the mechanisms that maintain STAT1 in a phosphorylation-competent conformation? Hsps not only function as molecular chaperones for newly synthesised proteins, but are also essential factors in the cell signalling pathways activated by inflammatory mediators.15 21 Hsp90 is a highly conserved and essential stress protein that functions as a positive regulator of cell signalling pathways by modifying or maintaining the conformation of its client proteins for active signalling. Moreover, blocking the ATPase site of Hsp90 using geldanamycin or its derivative 17-AAG inhibits the function of these client proteins.22 We previously showed that the binding of Hsp90 to its client protein IKKα/β may be necessary to maintain solubility and thus the function of this protein.15 Furthermore, a previous study reported that STAT3 is a client protein of Hsp90 and that this interaction is required for STAT3 phosphorylation.18 We found that STAT1 and Hsp90 form a complex under non-stimulated conditions, albeit a fraction of the total STAT1 population, and that this complex is disrupted after heat stress (figure 8). Furthermore, we show that STAT1 and Hsp90 are recruited to the plasma membrane under IFNγ-stimulated conditions. Thus, we postulate that in alveolar macrophages, Hsp90 binding to STAT1 is necessary for IFNγ-mediated STAT1 activation. Activated STAT1 is not detected in the insoluble fraction since it is functional to interact with other proteins in the signalling pathway. In contrast, STAT1 becomes insoluble after heat stress, is not phosphorylated by the kinases JAK1/2 and is degraded. Any IFNγ-mediated degradation of STAT1 is postactivation while the heat stress-mediated degradation due to protein unfolding is pre-IFNγ activation. From our studies, STAT1 protein can be added to the growing list of Hsp90 client proteins.23

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
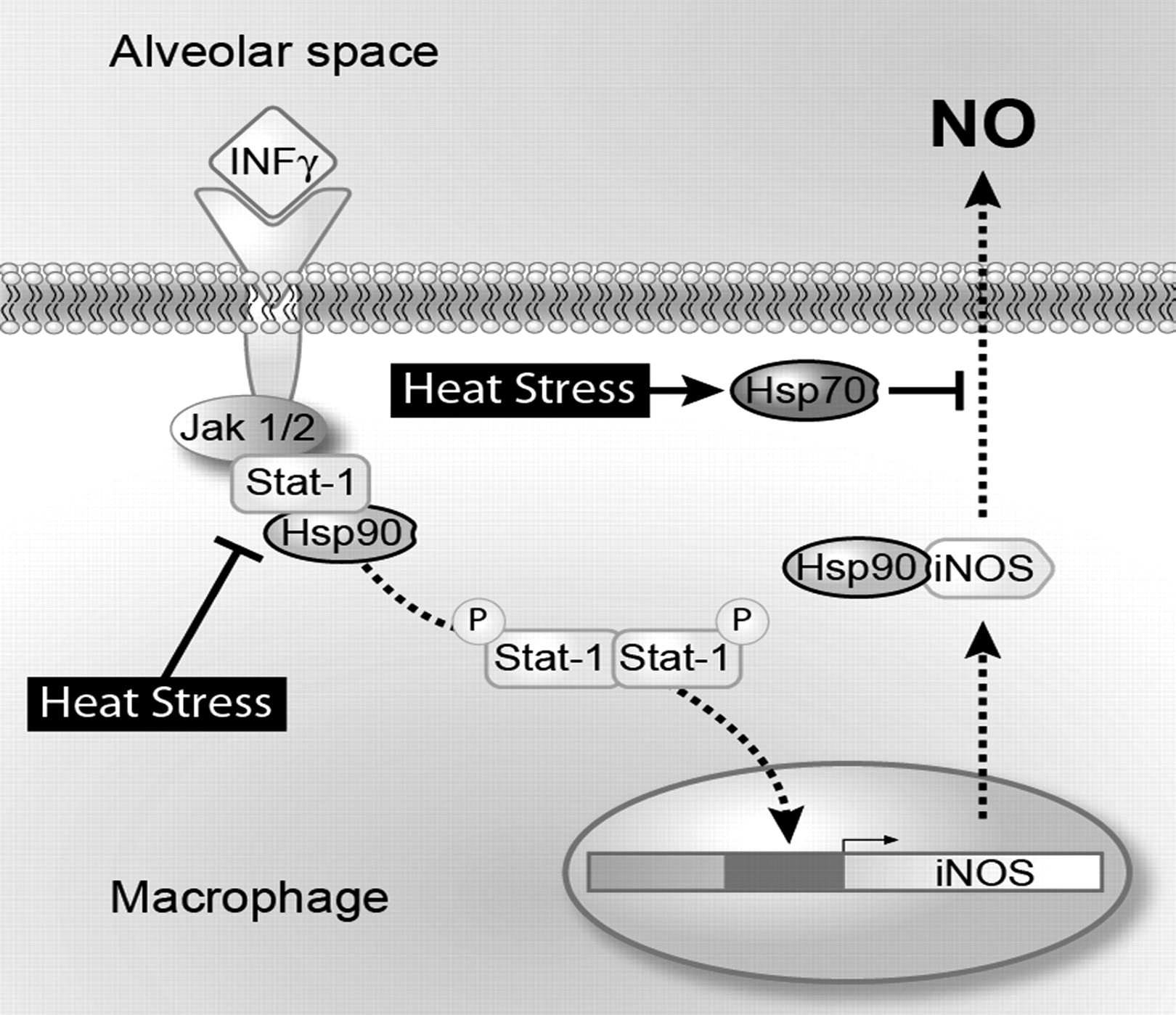{kind=link}
Schematic of heat stress inhibition of signal transducer and activator of transcription factor 1 (STAT1) and inducible nitric oxide synthase (iNOS) signalling pathways. Heat stress or treatment of macrophages with 17-allylamino-17-demethoxygeldanamycin (17-AAG) results in the disruption of the STAT1–heat shock protein (Hsp90) complex preventing STAT1 phosphorylation and translocation to the nucleus. Postheat stress when the STAT1 signalling pathway is responsive to interferon γ (IFNγ) stimulation, nitric oxide (NO) production from iNOS is inhibited due to the inducible form of Hsp70.
While the STAT1 signalling pathway recovers function 12 h postactivation of the SPR, the recovery of iNOS function takes 48 h. The iNOS protein has been shown to be a client protein of Hsp9024; however, the length of the recovery of iNOS function indicates that another mechanism besides the disruption of Hsp90–iNOS binding could also be involved in the inhibition of iNOS after heat stress (figure 8). Previous studies using overexpression of recombinant Hsp70i have shown that Hsp70i can inhibit iNOS function.25 26 However, the role of the SPR-mediated Hsp70i expression in modulating iNOS function is unknown. Our initial experiment showed that there was a 48 h recovery from SPR activation before alveolar macrophages would release NO in response to IFNγ stimulation. The NO release inversely correlated with the level of Hsp70i protein. Further investigation using either siRNA to Hsp70i to inhibit the heat stress-induced expression of Hsp70i or recombinant adenovirus to overexpress Hsp70i in the absence of SPR activation showed that it is Hsp70i expression that modulates iNOS function and NO production in alveolar macrophages and not biochemical changes due to heat stress. What is the mechanism of Hsp70i attenuation of iNOS-dependent NO release in alveolar macrophages? Some studies have shown that Hsp70i can inhibit iNOS expression.26–28 This does not appear to be the mechanism in alveolar macrophages since we can detect iNOS protein in the detergent-soluble fraction of these cells. In support of our findings, one recent study identified iNOS–Hsp70i complex formation in mouse intestinal tissue lysates and suggested that Hsp70i may regulate iNOS function.25 We are currently investigating possible mechanisms of attenuation of iNOS function by Hsp70i in alveolar macrophages. Our working hypothesis is that Hsp90 is a critical part of cell signalling pathways such as STAT1/iNOS that mediate lung fluid balance abnormalities induced by ALI. SPR activation may protect the integrity of the alveolar–capillary barrier by two mechanisms: (1) first by an immediate dissociation of Hsp90 from its client proteins rendering these proteins non-functional, then (2) by a delayed phase involving the de novo synthesis of Hsp70 that binds to and temporarily inhibits the function of some of the Hsp90 client proteins until Hsp90 can re-complex with these proteins.
What are the clinical implications of our findings? The airspace release of iNOS-dependent NO is deleterious during the early phase of ALI. The release of NO by alveolar macrophages and lung epithelial cells inhibits ion channel function via post-translational modification, thus preventing removal of oedema fluid from the airspace of the lungs.29 Several studies have reported that prior activation of the SPR prevents the abnormalities in the lung fluid balance associated with ALI.12 30 31 These studies may have an important therapeutic significance in humans. We recently reported that SPR activation occurs in patients with ALI and correlates with the preservation of AFC.9 Thus, the stress response could be activated as a prophylactic therapy to protect from lung fluid balance abnormalities associated with ALI, using pharmacological inhibitors of Hsp90 that have been shown to be safe in humans32 but do not induce Hsp70.
Acknowledgments
The authors thank Drs Kimberly Mace, Natasha Bir and Nancy Boudreau for critical review of the manuscript. This work was primarily supported by UCSF Academic Senate Grant (MH), NIH Grant GM 62188 (JFP), ALA Senior Research Training Fellowship and T32 GM008440 (JR).
References
Supplementary materials
Web Only Data thx.2008.101139
Files in this Data Supplement:
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.