Article Text

Abstract
Background Ventilator-associated pneumonia (VAP) is the most commonly fatal nosocomial infection. Clinical diagnosis of VAP remains notoriously inaccurate. The hypothesis was tested that significantly augmented inflammatory markers distinguish VAP from conditions closely mimicking VAP.
Methods A prospective, observational cohort study was carried out in two university hospital intensive care units recruiting 73 patients with clinically suspected VAP, and a semi-urban primary care practice recruiting a reference group of 21 age- and sex-matched volunteers.
Growth of pathogens at >104 colony-forming units (cfu)/ml of bronchoalveolar lavage fluid (BALF) distinguished VAP from “non-VAP”. Inflammatory mediators were quantified in BALF and serum. Mediators showing significant differences between patients with and without VAP were analysed for diagnostic utility by receiver operator characteristic (ROC) curves.
Results Seventy-two patients had recoverable lavage—24% had VAP. BALF interleukin-1β (IL-1β), IL-8, granulocyte colony-stimulating factor and macrophage inflammatory protein-1α were significantly higher in the VAP group (all p<0.005). Using a cut-off of 10 pg/ml, BALF IL-1β generated negative likelihood ratios for VAP of 0.09. In patients with BALF IL-1β <10 pg/ml the post-test probability of VAP was 2.8%. Using a cut-off value for IL-8 of 2 ng/ml, the positive likelihood ratio was 5.03. There was no difference in cytokine levels between patients with sterile BALF and those with growth of <104 cfu/ml.
Conclusions BALF IL-1β and IL-8 are amongst the strongest markers yet identified for accurately demarcating VAP within the larger population of patients with suspected VAP. These findings have potential implications for reduction in unnecessary antibiotic use but require further validation in larger populations.
- Assisted ventilation
- cytokine biology
- bacterial infection
- bronchoscopy
- pneumonia
Statistics from Altmetric.com
Introduction
Ventilator-associated pneumonia (VAP) occurs in up to 20% of patients mechanically ventilated for >48 h and is typically associated with mortality rates of ∼30%.1 2 VAP therefore has a higher mortality than any other hospital-acquired infection, and exerts a large financial burden on health services.3 Although estimates of the attributable mortality vary, most studies describe a significant associated mortality and morbidity.1 3 4
The optimal methods and criteria required to make a diagnosis of VAP remain contentious.1 However, it is generally accepted that only approximately a quarter to one-third of patients with clinically suspected VAP satisfy predefined microbiological criteria for pneumonia.5 6 The clinical diagnosis of VAP is made relatively frequently in the intensive care unit (ICU). This can result in empirical antibiotic treatment, with the inherent risk of overprescribing.7–9 Therefore, in recent years attempts have been made to identify biological markers that can distinguish VAP accurately, rapidly and practically.10–12
Given the key roles played by inflammatory cytokines and neutrophils in the natural history of pneumonia, it is plausible that discriminatory markers of infection may be found in the innate immune system. We hypothesised that in critically ill patients pneumonia would drive a further increase in pulmonary inflammation, with inflammatory mediators distinguishing VAP from conditions that mimic VAP.
Materials and methods
Patients
This prospective, observational, cohort study was performed in two university hospital general ICUs admitting all patients except those undergoing cardiothoracic surgery. Patients were screened daily for possible VAP, and were assessed for enrolment as soon as the clinical suspicion was raised. Where enrolment occurred, bronchoscopy took place within 6 h of clinical diagnosis. Demographic details, co-morbidities and prescribed medications were recorded. The severity of presenting illness was assessed by an Acute Physiology and Chronic Health Evaluation II (APACHE II) score calculated within 24 h of ICU admission. Patients were eligible if they fulfilled recognised criteria for clinically suspected VAP—that is, mechanical ventilation for at least 48 h, new and persisting infiltrates on a chest radiograph and at least two of the following: purulent tracheal secretions, temperature >38°C or white cell count >11×109/l—based on a modification of previously published clinical criteria.1 Exclusion criteria comprised PaO2 <8 kPa on FiO2 >0.7, positive end-expiratory pressure >15 cm H2O, active bronchospasm, myocardial infarction within the last 3 months, unstable arrhythmia, mean arterial pressure <65 mm Hg on vasopressor therapy, bleeding diathesis (including platelet count <20×109/l) and initiation or modification of antibiotics in the preceding 3 days.1 Patients who had had no change in prescribed antibiotics for >3 days were included.1 13
Patients had fibreoptic bronchoscopy and bronchoalveolar lavage (BAL) performed by a single experienced operator using a predefined, standardised technique.14 15 Briefly, where focal infiltrates were present, the bronchoscope was wedged in a subsegment corresponding to the area of radiological involvement. In the case of diffuse radiographic change the bronchoscope was wedged in a subsegment producing visible purulent secretions or (in the absence of purulent secretions) in the posterior segment of the right lower lobe. A 20 ml aliquot of sterile saline was instilled and the aspirate (representing a “bronchiolar” sample) discarded, then 200 ml of sterile saline was instilled in aliquots and the aspirate (representing an alveolar sample) retained. Whole blood was collected into 0.38% sodium citrate (final concentration).
Volunteer reference group
After recruitment of 40 patients the (anonymised) age and sex of each patient was communicated to a local primary care practice, where staff unconnected with the study randomly identified matching individuals and sent out invitations to participate. The first twenty-one respondents were enrolled to form a reference group. Exclusion criteria comprised hypoxia (SaO2 <92% on air), bleeding diathesis, anticoagulant therapy, insulin-dependent diabetes mellitus, arrhythmia, bronchospasm not responding to nebulised β2 agonist or clinical evidence of respiratory tract infection. Eligible volunteers provided blood and had fibreoptic bronchoscopy and BAL performed by the same investigator as above.
Processing of BAL fluid (BALF) and whole blood
An aliquot of BALF was sent to the National Health Service (NHS) Clinical Microbiology laboratory for culture, whilst simultaneous cultures were undertaken in our research laboratory. Samples were processed using a standard operating procedure (SOP) in accordance with the SOP for the processing of BAL issued by the UK Health Protection Agency (HPA).16 Analyses were limited to those routinely performed on BALF in our NHS laboratory (ie, detailed analysis for viruses was not included), with the exception that we additionally performed anaerobic cultures. BALF from healthy volunteers was only cultured in the research laboratory.
Growth of >104 colony-forming units (cfu) per ml of lavage fluid confirmed VAP.1 This definition is used by several infection control and critical care organisations including the Hospitals in Europe Link for Infection Control through Surveillance (HELICS) programme.17 Patients whose BALF grew <104 cfu/ml formed a “non-VAP” category. For further subgroup analysis “non-VAP” was subdivided into “sub-VAP growth” and “sterile” groups. A 1 ml aliquot of BALF was collected for culture, and the remainder centrifuged at 700 g for 10 min. Supernatant was immediately frozen at −80°C until further analysis. The cellular pellet was resuspended in warmed Iscove's modified Dulbecco's medium (IMDM; Invitrogen, Carlsbad, California, USA) and cytospins produced. Cytospins were stained with Diff-Quik (Reagena, Toivala, Finland) and differential cell counts established.
A 30 ml aliquot of citrated whole blood was separated into cellular and plasma components by centrifugation.18 Serum was prepared by adding 1 M calcium chloride to plasma.
Quantification of cytokines and inflammatory mediators
Concentrations of tumour necrosis factor-α (TNFα), interleukin (IL)-1β, IL-6, IL-8, IL-10, granulocyte colony-stimulating factor (G-CSF) and macrophage inflammatory protein-1α (MIP-1α) in serum and BALF were estimated using cytometric bead array (CBA) kits (BD Bioscience, Franklin Lakes, New Jersey, USA). The concentrations of type 1 soluble triggering receptor expressed on myeloid cells (sTREM-1) and monocyte chemoattractant peptide 1 (MCP-1) were measured by ELISA (R&D Systems, Minneapolis, Minnesota, USA). Samples measured by CBA and ELISA were diluted in an assay-dependent manner to ensure they lay within the limits of the calibration curves. The dilution required ranged from neat to 1:100 for the highest values. Urea was measured by a colorimetric method (QuantiChrom, Bioassay Systems, Hayward, California, USA) and specifically used as a recognised means of correcting for dilutional effects in BALF.19
Consent and ethics approval
Informed, witnessed assent was obtained from a relative or main carer for all patients. Informed, written consent was obtained from all volunteers. The study was approved by the relevant Research Ethics Committees.
Statistical analysis
Statistical analysis was conducted using Prism (Graphpad Software, San Diego, California, USA). Non-normally distributed data were analysed using the Mann–Whitney U test for two variables and the Kruskal–Wallis test for greater than two variables, using the Dunn method for posthoc analysis. Normally distributed data were analysed using the Student t test or analysis of variance (ANOVA) with the Bonferroni method for posthoc analysis. Preliminary identification of candidate biomarkers was undertaken by noting those with significant differences between the VAP and “non-VAP” median values. The diagnostic utility of these variables was assessed using area under the receiver operator characteristic (ROC) curves. For those with area under the curve values of ≥0.5, optimal cut-offs and likelihood ratios were determined by the value with the maximum Youden index20; a likelihood ratio is a likelihood that a person with a positive (or negative) test has the disease in question. For the two most promising candidates discriminating VAP from “non-VAP”, multilevel likelihood ratios were calculated to illustrate diagnostic potential. Combinations of measures were assessed for enhanced diagnostic potential by statistical modelling via logistic regression and classification tree methods.
Results
There were 74 eligible patients; 73 were enrolled, with one excluded due to lack of a relative's informed assent. Seventy-two patients had recoverable BALF and so entered the analysis. Seventeen (24%) grew organisms at >104 cfu/ml of BALF (“VAP group”) (growing a median of 5.7×104 cfu/ml, IQR 3×104–6×104 cfu/ml). Seven grew a Gram-positive organism, six a Gram-negative organism, and four patients grew fungi, including three yeasts (table 1). One patient growing a Gram-positive organism also grew anaerobes. The remaining 55 patients formed the “non-VAP” group, of whom 22 were categorised as “sub-VAP” (ie, organisms cultured but at <104 cfu/ml of BALF, growing a median 2.1×102 cfu/ml (IQR 7.5×102–4×102 cfu/ml)) and 33 as “sterile”. Comparisons between cultures conducted in the NHS laboratory and our laboratory revealed strong agreement, with all patients identified as having VAP by the research laboratory being similarly identified by the NHS laboratory. There were no discordant cultures in this group. Amongst the non-VAP group two patients grew bacteria (<102 cfu/ml) which were not reported by the NHS laboratory, and one sample grew bacteria (<102 cfu/ml) in the NHS laboratory but not the research laboratory. Twenty-one volunteers were recruited to form the reference group. Twelve samples grew mixed oral commensal flora but always at <102 cfu/ml.
Organisms grown in patients with and without ventilator-associated pneumonia (VAP)
In the VAP group 18 species were grown from 17 patients (ie, two organisms were grown from one patient); by definition organisms were grown at >104 cfu/ml BALF. In the non-VAP group 27 species were grown from 22 patients (ie, two organisms were grown from five patients); by definition organisms were grown at <104 cfu/ml BALF.
The three groups studied were closely matched with respect to age (table 2). The VAP and non-VAP groups were similar with regard to duration of mechanical ventilation, severity of illness and co-morbidities (table 2). Although there was a greater likelihood of patients in the VAP group being male, having a surgical reason for admission to ICU and having less acute lung injury (ALI)/adult respiratory distress syndrome (ARDS), these differences did not reach statistical significance (table 2).
Demographic and clinical details of patients and age-/sex-matched volunteers
All measured cytokines and inflammatory mediators in serum showed similar concentrations in the VAP and non-VAP groups (Supplementary table S1). No serum marker appeared to have potential value for discriminating VAP from non-VAP, though a trend in this direction was observed for sTREM-1. In general, serum markers were higher in both patient groups than in volunteers, with the exceptions of IL-1β and TNFα, which were broadly similar in all three groups.
In contrast, the VAP group had significantly higher concentrations of IL-1β, IL-8, G-CSF and MIP-1α in BALF than the non-VAP group (table 3). Trends in the same direction were observed for IL-6 and sTREM-1 (table 3).
Inflammatory profile of bronchoalveolar lavage fluid (BALF)
On the basis of these findings, the capacity for IL-1β, IL-8, G-CSF, MIP-1α, IL-6 and sTREM-1 to distinguish VAP among the population with clinically suspected VAP was tested (figure 1). For the ROC curves plotted in figure 1, area under the curve correlates with the discriminatory value of the marker being analysed. In this context IL-1β and IL-8 appeared to delineate VAP most accurately. When optimal cut-off values were derived, concentrations of BALF IL-1β <10 pg/ml appeared to be particularly powerful for the exclusion of VAP. Specifically, where BALF IL-1β was <10 pg/ml, the negative likelihood ratio of 0.09 gives a post-test probability of having VAP calculated at 2.8% (95% CI 0.1% to 15.9%). IL-1β proved less useful as a positive discriminator of VAP. In contrast, IL-8, whilst being less powerful for the exclusion of VAP, had a broader diagnostic value. Thus, relative to the identified cut-off value in figure 1 of 2000 pg/ml, a high BALF IL-8 concentration increased the post-test probability of VAP being present to 61.4% (95% CI 36.2% to 76.8%), derived from the positive likelihood ratio of 5.03. These data are presented as scatter plots in figure 2, illustrating that IL-1β has utility in excluding VAP, but yields a number of false positives. In contrast, IL-8 has a broader utility, but demonstrates a number of false-positive and false-negative values. Multilevel likelihood ratios demonstrated that increasing levels of BALF IL-8 lead to increased confidence for “ruling in” VAP, with the highest level examined (4000 ng/ml) producing a post-test probability of 75%. However, at this level, patient numbers are small and there is a marked reduction in sensitivity (details are presented in Supplementary table S2). A similar relationship was not demonstrated for IL1-β (see table S2). G-CSF, MIP-1α, IL-6 and sTREM-1 had markedly less discriminatory value than IL-1β or IL-8. Statistical modelling using combinations of mediators (via logistic regression and classification tree methods) failed to add discriminatory value to that achieved by either IL-1β or IL-8 alone. We did not find any difference in cytokine levels between VAPs caused by different classes of organism (Gram positive, Gram negative or fungi). Interestingly all cases associated with Candida albicans had cytokine levels above the optimum cut-off point for diagnosis for IL-1β, IL-8, G-CSF and MIP-1α.
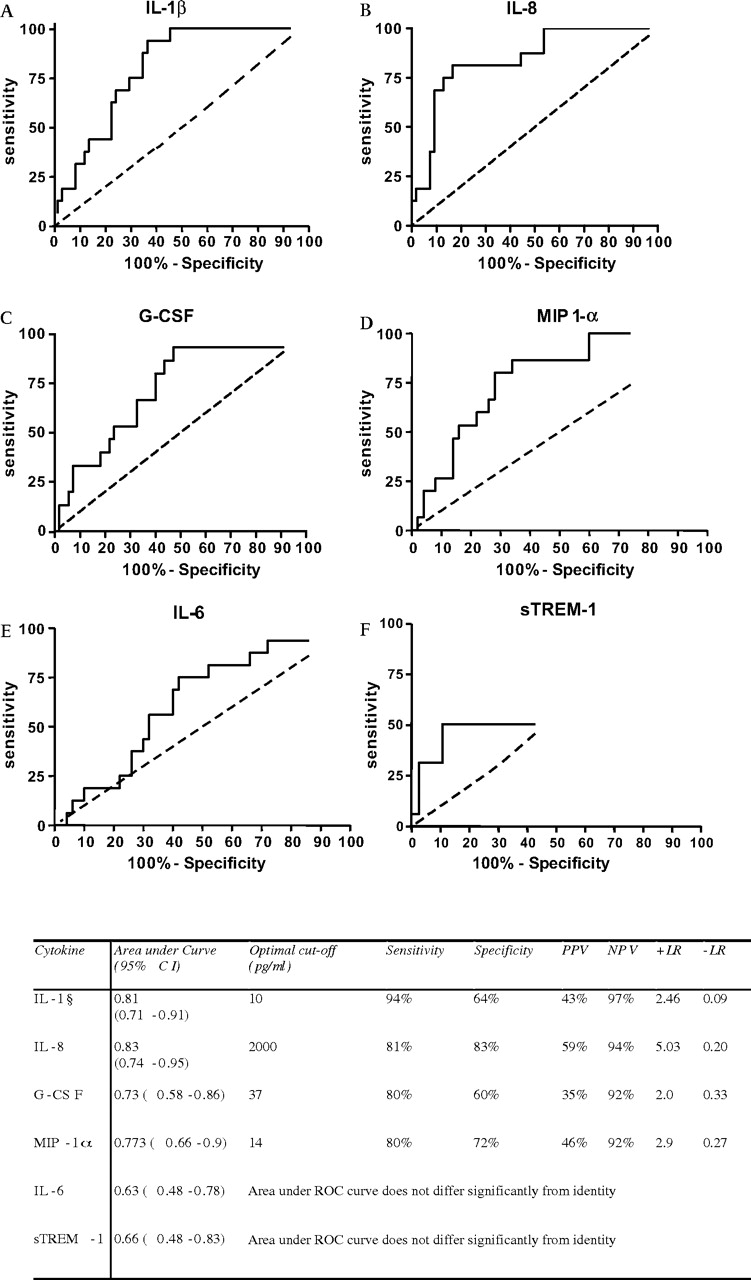
Receiver operating characteristic (ROC) curves and optimal sensitivity, specificity, positive predictive values (PPVs) and negative predictive values (NPVs) for bronchoalveolar lavage fluid (BALF) cytokines (n=72, 55 non-ventilator-associated pneumonia (VAP) and 17 VAP). Data are derived from the patients with clinically suspected VAP. The broken line shows identity. G-CSF, granulocyte colony-stimulating factor; IL, interleukin; +LR, positive likelihood ratio; −LR, negative likelihood ratio; MIP 1-α, macrophage inflammatory protein-1α; sTREM-1; type 1 soluble triggering receptor expressed on myeloid cells.

Scatter plots of pulmonary cytokine levels (n=72, 55 non-ventilator-associated pneumonia (VAP) and 17 VAP). Each dot represents a single observation. The solid lines mark the median values; the hashed line marks the optimal diagnostic cut-off. A log scale is used due to the skewed nature of cytokine levels. G-CSF, granulocyte colony-stimulating factor; IL, interleukin; MIP1-α, macrophage inflammatory protein-1α;
To examine the specificity of our findings, we analysed the cytokine levels in the non-VAP group subdivided into those with no growth (“sterile”) and those with growth below the 104 cfu/ml cut off (“sub-VAP growth”). Cytokine concentrations did not differ significantly between the sub-VAP and sterile groups (figure 3). No correlation was found between bacterial growth and cytokine concentration when examining those patients with growth below the 104 cfu/ml cut-off (r=0.14, p=0.31 for IL-8; r=0.06, p=0.65 for IL-1β by Spearman rho).

{kind=link}
{kind=link}
{kind=link}
Comparison of pulmonary cytokine levels between patients with ventilator-associated pneumonia (VAP) (n=17), patients with growth of pathogens in bronchoalveolar lavage fluid (BALF) below the diagnostic 104 cfu/ml cut-off (n=22), and patients with no growth in BALF (n=33). (A) Interleukin-8 (IL-8) levels. Data are presented as the median and IQRs, p<0.0001 by Kruskal–Wallis; NS (non-significant), p>0.05, **p<0.01,***p<0.0001 by the Dunn posthoc test. (B) IL-1β levels. Data are presented as the median and IQRs, p=0.0006 by Kruskal–Wallis; NS, p>0.05, **p<0.01,***p<0.0001 by the Dunn posthoc test.
Discussion
These data have implications both for the diagnosis of VAP and for understanding the biology of the disease. The practical implications are that, in our hands, a patient with a BALF IL-1β concentration <10 pg/ml has an ∼3% probability of having VAP. In contrast, the probability of VAP being present increases as the BALF IL-8 concentration increases; a level >2 ng/ml corresponding to a 61% probability of VAP being present.
VAP remains common and associated with a high mortality.1 4 It is difficult to diagnose accurately on clinical grounds alone,5 7 8 with a tendency to overdiagnosis leading to overprescription of antibiotics.1 7 8 Diagnostic confirmation remains largely reliant on standard microbiological culture techniques which generally take 24–48 h to yield results.15 In this regard, the capacity of IL-8 to increase the likelihood of a correct diagnosis fivefold and/or the capacity of low BALF IL-1β to reduce the likelihood of VAP 10-fold is an important development. The assays used here can yield a result within 4 h. These tests could therefore have a significant impact on clinical decision making.
These results also provide an interesting perspective on the biology of VAP. For the majority of markers both patient groups had higher levels than volunteers, implying a proinflammatory state in critically ill patients as might be expected. However, it is intriguing that acute inflammatory cytokines such as IL-1β and MIP-1α were so similar when comparing the non-VAP group with volunteers. Although non-significant, there was a trend towards more cases of ARDS/ALI in the non-VAP group. One might have anticipated that these (ARDS/ALI) patients would also demonstrate elevated cytokine levels21; however, we found no difference between IL-8 and IL-1β levels within the non-VAP group when subdivided into those with ARDS/ALI and those without. We believe this relates to the time point of sampling, with our patients' median length of stay prior to being recruited being 8 days.
In contrast, the VAP group appeared to have a brisk inflammatory response confined to the lung. We have no data relating to the source of the relevant cytokines, but diffusion from serum seems unlikely, suggesting a prominent role for alveolar epithelium and/or lung macrophages. It is tempting to speculate that sufficient pathogens in the alveolar space drive a renewed and compartmentalised inflammatory response. However, this study was not designed to address this specific question and we cannot exclude the possibility that VAP arises in those patients who first develop an upregulation of inflammation in the lung.
A number of potential biomarkers for VAP have been proposed, including sTREM-1, procalcitonin, copeptin and adrenomedullin.10–12 22 23 However, only sTREM-1 has been found to have a stronger negative predictive value (or area under the ROC curve) for VAP than the findings presented here,10 and recent studies have found less impressive predictive capabilities for sTREM-1 in BALF,24 25 in line with our observations. It is interesting to consider why IL-1β and IL-8 have not been identified as potential discriminatory markers in previous studies. The compartmentalised nature of proinflammatory cytokines in VAP has been noted previously,26 but the diagnostic capability observed in our study was not seen. We believe this discrepancy relates to the timing of investigation and reliance on rigorously collected alveolar samples in our study.
We recommend caution when considering the general applicability of our data. First, our results were generated using bronchoscopy and lavage which may be contraindicated in some patients, and remains relatively labour intensive and operator dependent.27 An ideal diagnostic marker for VAP would involve a highly discriminatory blood test. However. this remains elusive, as reflected in Supplementary table S1. Debate continues as to the optimal method for diagnosing VAP.28 The “gold standard” for the diagnosis of pneumonia remains histology29; however, this is neither desirable nor practicable in critically ill patients. Therefore, uncertainty over the optimal method of diagnosis affects all studies concerning VAP, especially those focusing on diagnostic markers. A strength of our study lies in the fact that we only recruited patients with predefined clinically suspected VAP using established criteria,1 17 and employed a rigorously standardised, visually directed BAL procedure that was conducted by a single experienced operator and relied on quantitative cultures to confirm or refute VAP. To our knowledge these specific, strict criteria have not been simultaneously applied in previous studies seeking diagnostic markers for VAP. Our incidence of confirmed VAP (24%) is at the lower end of the reported incidence, but is consistent with previous reports,30 and the 95% CI (14% to 34%) for the true population incidence overlaps with the estimate from several other reports.31 32 The precise timing of sampling may also influence the diagnosis of VAP. Our data relate specifically to the time of first clinical suspicion of VAP, and are based on the use of a particular assay system. Therefore, while IL-1β and IL-8 are promising diagnostic markers, their diagnostic usefulness (and the derived cut-off values) should be validated in wider populations of ICU patients.
Secondly, there was a non-significant trend towards more “non-VAP” patients being on antibiotics at the time of bronchoscopy. Antibiotics could have suppressed microbial growth to some degree in these patients, although previous studies suggest that after 3 days of unchanged antimicrobial treatment false-negative cultures are unusual.1 13 Nevertheless, this does not deflect the fact that marked rises in IL-1β and IL-8 only occurred in patients with >104 cfu/ml (figure 3). This suggests that low levels of bacterial growth may be tolerated in the alveolar space and support the concept of clinical VAP emerging above a bacterial threshold.1 15
Thirdly, the use of non-ventilated volunteers in this study can be questioned. We would stress that this group was recruited as a reference group only. The important question in clinical practice is whether patients with a high clinical suspicion of VAP have the condition or not; hence our critical comparison was between the VAP and non-VAP groups.
A fourth caveat relates to the variable case mix and microbiological epidemiology of ICUs.33 We included a wide variety of surgical and medical cases, but no cardiothoracic cases were involved. The range of bacteria isolated in this study broadly reflects those described elsewhere, but is notable for the absence of Pseudomonas aeruginosa. It may also be potentially relevant that we did not culture for respiratory viruses. Finally, the inclusion of fungi, especially Candida species, as causative organisms in VAP remains controversial.1 34 35 However, we analysed our data strictly in accordance with our predefined diagnostic definitions which were based on the concentration of organisms (without specifying the type). Interestingly we did not find any difference in cytokine levels between fungal and bacterial VAPs, with both demonstrating a profound and characteristic pulmonary inflammation. In our patients with VAP associated with fungal growth, all four cytokines described in figure 1 were above the optimal cut-off levels defined by the ROC curves. While we acknowledge that the small numbers involved mean that statistical non-significance could reflect a type two error, the data suggest that fungal infection was associated with the same pulmonary inflammation as the bacterial pathogens more typically associated with VAP. Furthermore, exclusion of C albicans and/or coagulase-negative staphylococci (sometimes considered “non-pathogenic” organisms) had no significant effect on the diagnostic usefulness of the cytokines as assessed by area under the ROC curve (data not shown).
In conclusion, VAP is associated with increased pulmonary IL-1β and IL-8. Further studies are warranted to validate IL-1β and IL-8 as diagnostic markers able to influence important clinical endpoints such antibiotic prescribing in ICUs.
Acknowledgments
The authors would like to acknowledge the help and support of Ms Pam Ramsay, Mr Gordon Mills and the staff of the Intensive Care Units of the Royal Infirmary of Edinburgh and Western General Hospital, Edinburgh.
References
Footnotes
Linked articles 127308.
Funding Sir Jules Thorn Charitable Trust, 24 Manchester Square, London W1U 3TH. DJD is a Wellcome Trust Career Development Fellow (Fellowship # 078265). Other Funders: Wellcome Trust.
Competing interests ACM has received an academic prize (travel and accommodation to attend an international conference) funded by Eli Lily. AJS has received expenses from Astra Zeneca and Glaxo Smith Kline (for travel and accommodation) to attend international educational conferences. IFL has received expenses from Astra Zeneca (for travel and accommodation) to attend international educational conferences. TSW is the recipient of an unrestricted educational grant from Wyeth Pharmaceuticals for work concerning epidemiology of ICU-acquired infection. All other authors have no conflicts of interest to declare
Ethics approval This study was conducted with the approval of the Lothian Research Ethics Committee.
ACM collected and analysed data, drafted the manuscript and approves the final version. KK conceived the study, collected and analysed data, drafted the manuscript and approves the final version. TSW collected and analysed data, reviewed the manuscript and approves the final version. OLMN collected and analysed data, reviewed the manuscript and approves the final version. KD collected and analysed data, reviewed the manuscript and approves the final version. LF collected data, reviewed the manuscript and approves the final version. TSW identified patients, assisted in data collection reviewed the manuscript and approves the final version. SJM identified patients, assisted in data collection reviewed the manuscript and approves the final version. DGS identified patients, assisted in data collection reviewed the manuscript and approves the final version. PJA identified patients, assisted in data collection reviewed the manuscript and approves the final version. NA analysed the data, reviewed the manuscript and approves the final version. JRG collected the data, reviewed the manuscript and approves the final version. IFL collected the data, reviewed the manuscript and approves the final version. HR identified the matched volunteers, reviewed the manuscript and approves the final version. DJD analysed the data, reviewed the manuscript and approves the final version. CH conceived the study, obtained funding, reviewed the manuscript and approves the final version. JMS conceived the study, obtained funding, reviewed the manuscript and approves the final version. AJS conceived the study, obtained funding, collected and analysed the data, drafted the manuscript and approves the final version.
Provenance and peer review Not commissioned; externally peer reviewed.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Linked Articles
- Editorial
- Airwaves
- Correction