Article Text

Abstract
Background: Osmotic agents, such as inhaled dry powder mannitol, may increase mucociliary clearance by rehydrating the airway surface liquid and thus act as disease-modifying treatments in cystic fibrosis (CF). This is the first therapeutic trial of inhaled mannitol in children with CF; it was compared with recombinant human deoxyribonuclease (rhDNase), the current best established mucolytic treatment.
Methods: 38 children were recruited to an open crossover study. Subjects underwent an initial bronchial provocation challenge with dry powder mannitol. Those children with a negative challenge were randomly allocated to one of three consecutive 12-week treatment blocks (inhaled mannitol alone, nebulised rhDNase alone and mannitol + rhDNase). The primary outcome was forced expiratory volume in 1 s (FEV1). A number of secondary outcome measures were also studied.
Results: Twenty children completed the study. Bronchoconstriction and cough associated with mannitol administration contributed to the high attrition rate. The mean increase in FEV1 following 12 weeks of treatment was 0.11 litres (6.7%) (p = 0.055) for mannitol alone, 0.12 litres (7.2%) (p = 0.03) for rhDNase alone and 0.03 litres (1.88%) (p = 0.67) for rhDNase and mannitol. None of the secondary clinical outcomes was statistically significantly different between treatments.
Conclusions: Inhaled mannitol was at least as effective as rhDNase after 3 months treatment. There was a marked individual variation in tolerance to mannitol and in response to treatment however. Children who do not respond to rhDNase many benefit from a trial of inhaled mannitol. The combination of mannitol and rhDNase was not useful.
Trial registration number: NCT00117208
Statistics from Altmetric.com
Many processes contribute to airway obstruction and progressive lung damage in cystic fibrosis (CF), including the production of abnormally viscid mucus, mucus hypersecretion and defective mucociliary clearance (MCC). Previous therapeutic approaches aimed at improving airway clearance have included suppression or eradication of bacterial infection and attenuation of inflammation. Mucolytic agents have also been used to reduce sputum viscosity. Recombinant human deoxyribonuclease (rhDNase) is a now a well established and widely used adjunctive treatment for CF. DNA derived from neutrophil degradation is present in very high concentrations in the sputum of patients with CF and is a major contributor to the viscosity of airway secretions. rhDNase cleaves this extracellular DNA, improving mucus rheology. Several studies have now clearly demonstrated improvements in lung function and fewer chest exacerbations in patients treated with rhDNase.1 2 3 It may also reduce neutrophilic airway inflammation.4 There is widespread variation in the individual responses to rhDNase,3 5 however, and a significant number of patients derive no clinical benefit.
Epithelial airway surface liquid (ASL) volume regulation is critical for normal MCC, and airway dehydration is now thought to be a key initiating event in the pathogenesis of CF lung disease.6 7 8 9 There has been recent interest in osmotically active agents which may potentially increase MCC by rehydrating the ASL and thus act as disease-modifying treatments in CF. Two recent studies have demonstrated a modest increase in forced expiratory volume in 1 s (FEV1) and fewer pulmonary exacerbations in patients with CF treated with nebulised hypertonic saline (HS).10 11 Inhaled dry powder mannitol represents another novel osmotic agent. Mannitol is a naturally occurring sugar alcohol which is stable as a dry powder and suitable for encapsulation. It has been widely used as a bronchial provocation agent in measuring bronchial responsiveness.12 13 14 A number of studies have also demonstrated a beneficial effect of inhaled mannitol on MCC in patients with bronchiectasis and CF.15 16 17 18 To date there has been one study examining the effect of mannitol in lung function in CF.19 This short-term safety and efficacy trial reported that it was well tolerated and improved lung function over a 2-week period. The current study represents the first therapeutic trial of dry powder mannitol in children with CF. We have compared it with rhDNase, the current best established mucolytic treatment, and tested the hypothesis that inhaled mannitol, alone or in combination with rhDNase, is as effective as rhDNase in improving respiratory function in children with CF.
Methods
Study population
Children with a diagnosis of CF on standard criteria20 were recruited from two institutions (The Royal Brompton & Harefield NHS Trust, London, UK and Great Ormond Street Hospital for Children NHS Trust, London, UK). Inclusion criteria were age between 8 and 18 years, the ability to perform repeatable reliable spirometry (according to American Thoracic Society (ATS) criteria21) and either currently receiving rhDNase treatment or having an FEV1 of >40% and <70% predicted normal value (and therefore judged eligible to receive rhDNase). Exclusion criteria were known hypersensitivity to mannitol, rhDNase or bronchodilators, sputum infection with methicillin-resistant Staphylococcus aureus (MRSA) or Burkholderia cepacia, portal hypertension or varices, significant haemoptysis (>60 ml) in the previous 12 months, breast feeding or pregnancy. In addition, in order to ensure clinical stability at enrolment, patients had to have no changes in treatment or new respiratory symptoms in the 2 weeks prior to study entry.
The study protocol was approved by the local research ethics committee, and full written carer and child age-appropriate consent was obtained prior to trial entry. The trial was registered with ClinicalTrials.gov (identifier NCT00117208).
Study design and procedures
We conducted a prospective, randomised, open-label, crossover study. All subjects underwent a bronchial provocation challenge with dry powder mannitol prior to study entry.22 Children were pretreated with bronchodilator, following which they received incrementally increasing doses of mannitol up to a maximum cumulative dose of 475 mg. FEV1 was measured at the start of the challenge and after each increase in dose. A positive challenge was defined as a fall of ⩾15% in FEV1 from baseline. These children were withdrawn from the main study. Subjects with a negative challenge were randomly allocated to one of three consecutive 12-week treatment blocks (mannitol alone, rhDNase alone and a combination of mannitol and rhDNase). There was a 2-week washout period during which patients already on rhDNase treatment discontinued it. In order to prevent a carry-over effect between treatment periods there was also a 2-week washout period between treatment blocks. Previous studies have shown that 2 weeks is sufficient washout for the effects of rhDNase to disappear.3 5 Any longer than this may have led to a clinical deterioration related to stopping mucolytic treatment in those patients in whom it was established treatment prior to study entry. All other concomitant treatment was continued during the study.
Spray-dried dry powder mannitol was prepackaged into gelatin capsules (40 mg per capsule) and administered via a breath-activated inhaler device (Osmohaler; Plastiape, Osnago, Italy). Patients were instructed on inhaler technique, and all manoeuvres during study visits were supervised by the study investigator. Subjects inhaled 400 mg (10 capsules) twice a day. All children were pretreated with their usual bronchodilator (400 μg of Salbutamol or 1 mg of Terbutaline) 15 min prior to inhalation of the mannitol. A 2.5 mg dose of rhDNase (Pulmozyme, Roche Products, Welwyn Garden City, UK) was administered through the patient’s normal nebuliser device twice a day.
The total study period was 41 weeks, subjects attending for seven visits. They were assessed at the start and end of each 12-week treatment period. Bronchodilators and physiotherapy were withheld for 4 h prior to each visit.
Outcome measures
The protocol-defined primary outcome was FEV1. Secondary outcome measures were forced vital capacity (FVC) and forced expiratory flow (FEF25–75), frequency of pulmonary exacerbations, sputum microbiology, exercise tolerance, quality of life scores and adverse events. At every visit, lung function was measured in accordance with ATS guidelines21 using a hand-held compact spirometer (Vitalograph 2120, Vitalograph, Buckingham, UK), which was calibrated with a 1 litre syringe prior to each visit. Flow volume loops were inspected and confirmed as satisfactory, and three measurements were taken to ensure repeatability. To minimise the effect of normal diurnal variation in lung function, >70% of study visits were at the same time of day.
Exercise tolerance was assessed by subjects performing a 3 min step test at each visit. Standard age-appropriate validated quality of life (CFQ-R) questionnaires were completed by the children and additionally a parent/guardian in those under 14. Subjects also completed respiratory symptom and treatment effect questionnaires at each visit. Sputum was collected for microbiology at the start and end of each treatment block from all those children able to expectorate spontaneously. Cough swabs were taken for the remaining children.
Information on adverse events was collected at every visit from the patient-held study diaries given to all participants at the start of the study. Treatment adherence was assessed by asking all subjects to return unused mannitol capsules and empty vials of rhDNase.
Statistical analysis
A sample size of 42 children would allow a mean difference in FEV1 of 0.1 litres (∼8%) between treatments to be detected with 90% power at the 5% significance level, as long as the standard deviation of the treatment difference did not exceed 0.2 litres. Allowing for subject withdrawals, in order to have 42 children completing all three treatment periods (and therefore to be included in the per-protocol analysis), we aimed to recruit a total of 48 patients.
A prespecified per-protocol statistical analysis plan was followed and applied to the data from those children who completed all three treatment periods. The analysis focused on two separate pairwise comparisons of the treatments: mannitol alone versus rhDNase alone, and mannitol plus rhDNase together versus rhDNase alone.
A regression model compared within-subject differences between outcomes at the end of each treatment period (Y1–Y0), with adjustment for differences in the same outcome measure at the beginning of these periods (X1–X0). The regression equation is: (Y1–Y0) = a+b(X1–X0) where a, the intercept term, is the adjusted difference in mean outcome between the two treatments being compared. The ordinary least squares method was used to estimate a and b; the assumption of normally distributed residuals was investigated graphically for each model and found to be satisfied.
FEV1 and the secondary outcome measures were analysed on a measurement scale and the results presented as mean differences. In addition, the primary outcome, FEV1, was analysed on a log scale, and the results presented as percentage differences between treatments. Pulmonary exacerbations were compared between treatments using McNemar’s test. A p value <0.05 was considered statistically significant throughout.
Results
Forty-five children were recruited to the study, 38 of whom underwent an airway challenge with inhaled mannitol. Seven were not enrolled due to clinical instability. Nine children had a positive challenge test with mannitol. One further challenge was abandoned due to nausea. Twenty-eight subjects were therefore included in the main study and randomly allocated to a treatment sequence. Eight children withdrew during the study. The primary reason sited was troublesome cough in 6/8 (in 3 of whom there was evidence of a chest exacerbation at the time of withdrawal), nausea in 1/8 and treatment burden in 1/8. Treatment burden was reported as a contributory factor in a further 2/8 patients in whom cough was the main problem. The per-protocol analysis presented here is based on the 20 children who completed all three treatment periods (fig 1).
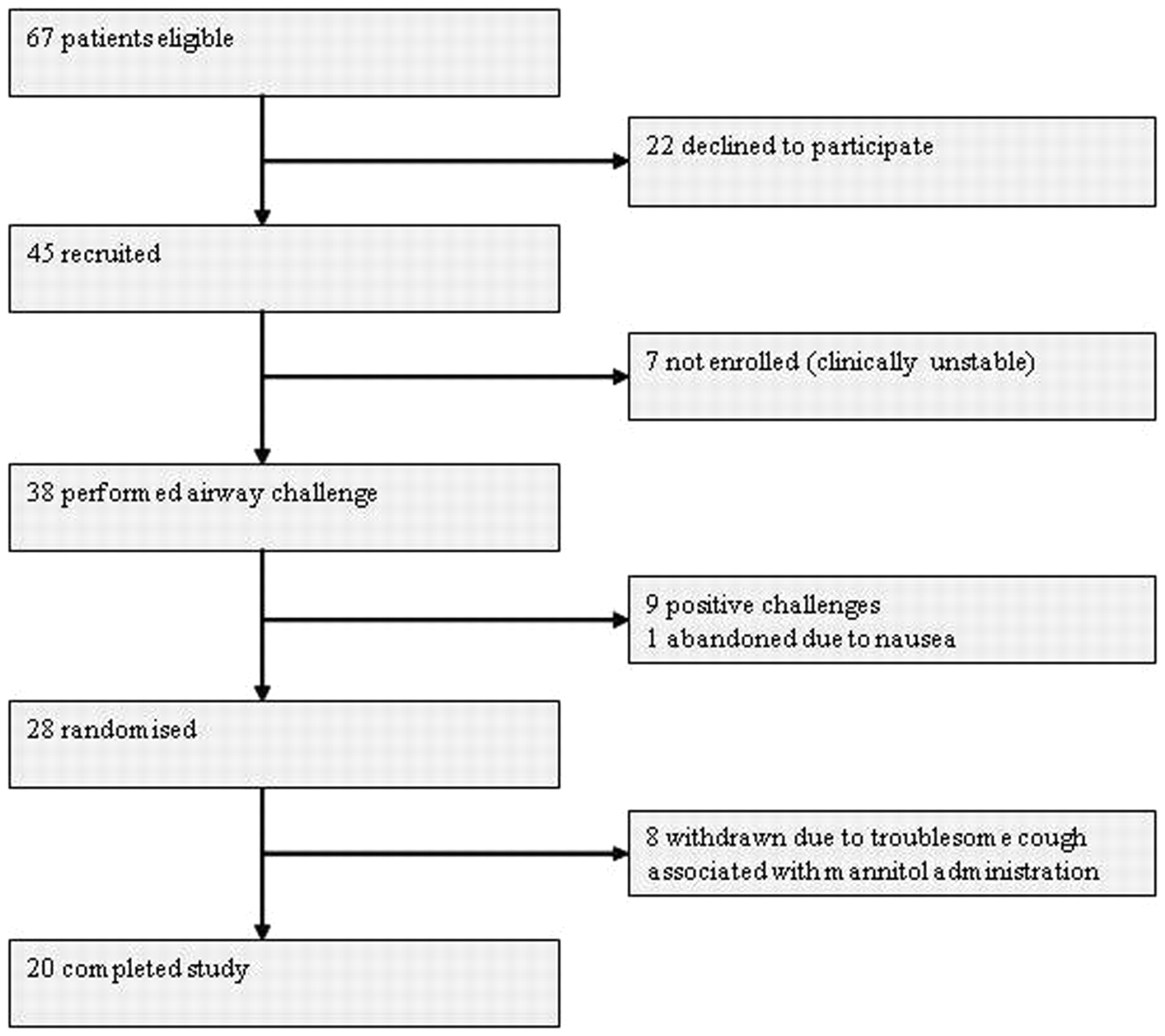
Trial profile.
The mean age was 13.2 years (SD 2.4 years), and 14 (70%) were girls. The mean baseline FEV1 was 1.67 litres (SD 0.50 litres) (mean 64% of predicted FEV1; SD 10%) (table 1).
Baseline characteristics of the study population
The adequacy of the washout periods in preventing carry-over was assessed by comparing, in turn, the pretreatment assessments for periods two and three with that for period one. We found no evidence of carry-over effects between treatment periods.
Nineteen children returned treatment packs and vials. Adherence was >70% in 14/19 patients with mannitol, 13/19 with rhDNase and 17/19 with a combination of both treatments. There was, therefore, no statistically significant difference in adherence in the different treatment groups.
The mean percentage change in FEV1 over each treatment period (final FEV1 minus initial FEV1 expressed as a percentage of initial FEV1) is shown in table 2.
Comparison of mean percentage differences in FEV1 between treatments over the 12 week treatment period
The mean FEV1 between the start and end of the treatment period increased with mannitol alone (0.11 litres (6.7%), 95% CI 0.00 to 0.23, p = 0.055) and with rhDNase alone (0.12 litres (7.2%), 95% CI 0.01 to 0.22, p = 0.03), but not with mannitol and rhDNase combined (0.03 litres, 95% CI −0.12 to +0.18, p = 0.67). The relative effects of treatments were also calculated as the mean difference in post-treatment outcome, adjusted for any start of treatment period difference. Comparing mannitol alone with rhDNase alone, there was a non-statistically significant trend for a higher FEV1 (0.05 litres, 2.8%) with mannitol (95% CI −4.2% to 10.4%, p = 0.42).
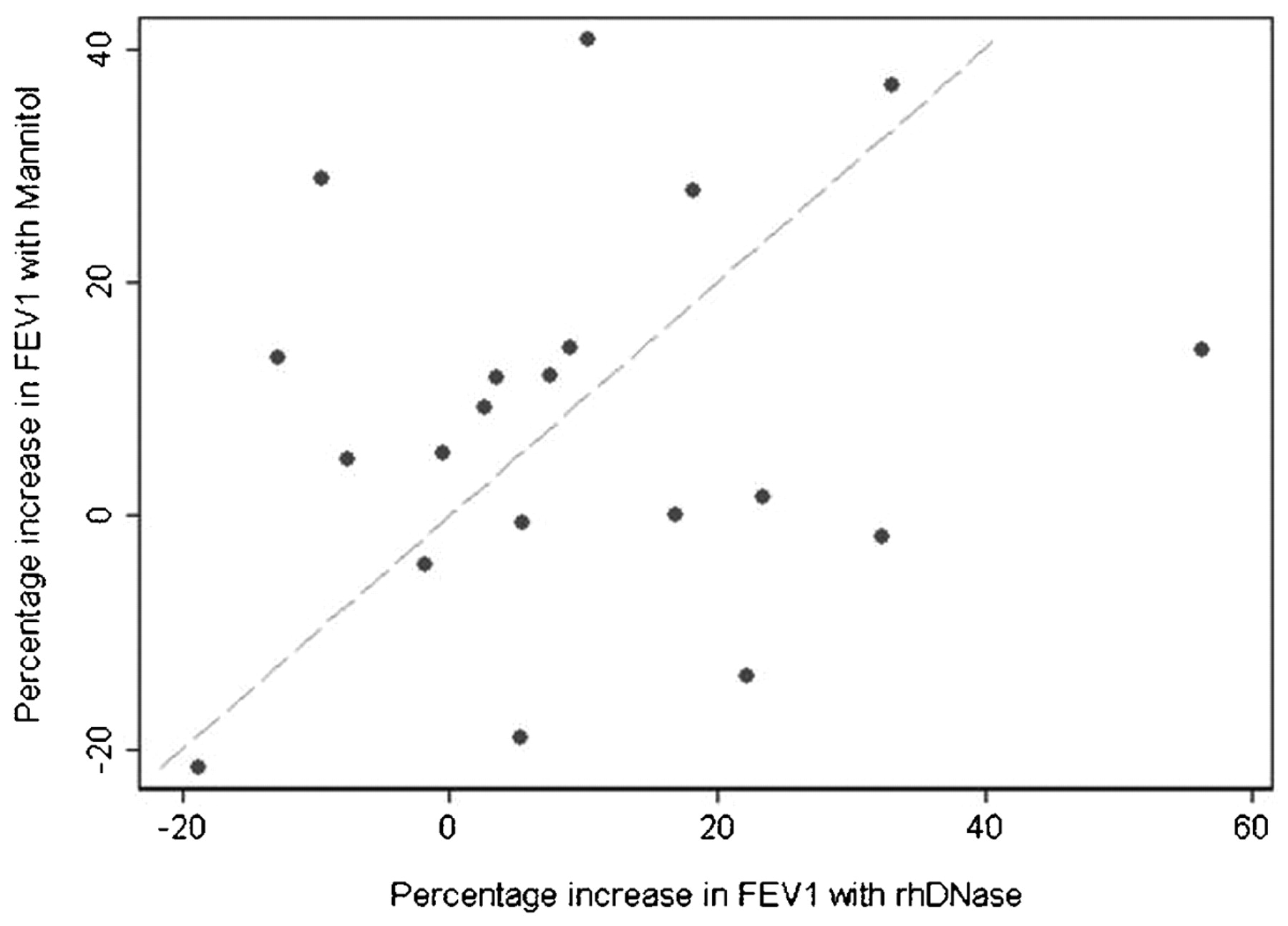
We found marked variation between individuals in response to mannitol and rhDNase, and the percentage change in FEV1 with mannitol alone and rhDNase alone for individual children is shown in fig 2.

{kind=link}
{kind=link}
Individual patient’s changes in forced expiratory volume in 1 s (FEV1) (% changes in FEV1 with recombinant human deoxyribonuclease (rhDNase) vs % changes in FEV1 with mannitol).
Whilst an improvement in FVC was apparent following treatment with rhDNase alone and mannitol alone, there was evidence of a trend towards greater improvement with mannitol (p = 0.053, table 3).
Mean percentage change in FVC and FEF25–75 between treatments, adjusted for baseline
There was no change in FVC following treatment with mannitol and rhDNase. There was no evidence of a difference between treatment groups for any of the other secondary outcome measures.
Discussion
We report that 3 months treatment with inhaled mannitol was as effective as rhDNase (the current best established mucolytic treatment), with each treatment resulting in a 7% increase in FEV1 from baseline. The combination of the two treatments was not useful. There was marked individual variation in response to both treatments with wide confidence intervals for the increase in FEV1. Nine of 20 children had an improvement of >10% in FEV1 with mannitol and 8/20 with rhDNase, undoubtedly clinically significant improvements. Figure 2 also suggests that patients who do not respond to rhDNase may derive benefit from inhaled mannitol. The three patients with the largest response to a combination of mannitol and rhDNase had modest responses to the individual treatments. There was no statistically significant difference in any of the secondary outcome measures. In particular there was no evidence of increased growth of Pseudomonas aeruginosa in sputum in the mannitol-treated group.
This study represents the first therapeutic trial of mannitol in children with CF. Bronchoconstriction and cough significantly reduced the tolerability of mannitol, and this contributed to the high study attrition rate (47%) and smaller numbers completing the study. Nine of 38 (24%) subjects were withdrawn due to a positive airway challenge. The majority of patients reported significant cough during administration of mannitol, both with the initial airway challenge and with subsequent regular administration. Six of the eight children who subsequently withdrew from the study sited cough associated with administration of mannitol as the main reason. Both these problems occurred despite pretreatment with bronchodilators. The study achieves 61% power with 20 children completing the study.
A comparative therapeutic trial would ideally adopt a double-bind design. This was impossible in the current study as the presentation and mode of delivery of the two treatments is very different. Mannitol is a dry powder packaged in gelatine capsules which are inhaled via a breath-activated inhaler device (Osmohaler). It tastes sweet and is administered twice a day. rhDNase is inhaled as an aerosol delivered via a nebuliser device once a day. There has been much debate on the appropriateness of a crossover design to examine an intervention that potentially affects the natural history of a disease.23 24 25 Two recent trials of azithromycin and rhDNase treatment in children with CF have successfully employed a crossover design.3 23 The main disadvantage with crossover trials is related to potential treatment interactions over the study period, and in particular carry-over effects. This is overcome by including sufficiently long washout periods between treatment blocks. There was no evidence of carry-over effect in the current study.
Outcome measures in trials of CF treatment are an area of great interest currently.26 27 28 FEV1 currently remains the best established and most objective measure of clinical benefit associated with a therapeutic intervention in CF. It might not be the most appropriate outcome measure for a potentially bronchoconstricting treatment however. Alternatives such as lung clearance index (LCI) or radiolabelled measures of MCC may need consideration in future trials.
An upper limit for FEV1 of 70% predicted value was chosen for two main reasons. First, a baseline FEV1 approaching 100% of predicted value is unlikely to allow a clear statistically measurable change in FEV1 associated with each treatment regimen. Secondly, in order to justify commencement of rhDNase treatment, it was felt that patients should have at least moderate lung disease (reflected in the chosen cut-off of 70%). Recently, rhDNase has been increasingly used in patients with milder chest phenotypes,29 and an increase in the upper limit for baseline FEV1 may be justifiable. With a treatment that potentially causes acute bronchoconstriction, a safe lower limit for FEV1 of 40% of predicted value was chosen.
The dose of mannitol utilised for the airway challenge has been previously reviewed.22 It may be that utilising a slightly lower cumulative dose of mannitol for the airway challenge will allow more children to be recruited to a therapeutic trial. Similarly the therapeutic dose of mannitol employed in the current study is based largely on data from studies in adults. The pilot study on MCC in adult patients with CF used 300 mg of mannitol,18 and the recent short-term safety and efficacy study used 420 mg.19 More recent evidence in adults with bronchiectasis suggests there is a dose response to at least 480 mg.30 It may be that, in children, a lower dose may have resulted in less coughing and improve adherence.
The increase in FEV1 with rhDNase is similar to that reported previously3 and that for mannitol was approximately equivalent. The lack of evidence for an increase in sputum infection with P aeruginosa is important as it has been suggested that mannitol could theoretically act as a bacterial energy source. The initial pilot study reported mannitol was safe and well tolerated.19 The current trial suggests that it may cause more bronchoconstriction and troublesome cough than previously reported. Jacques et al also suggested that a combination of mannitol and rhDNase may be better than either treatment alone. This is not supported by our findings, but larger numbers are required to study this effect further. We can only speculate as to why the combination of the two mucolytics was not beneficial. Our results suggest adherence is not an issue, although it is possible that the burden of two inhaled therapies resulted in less than a full treatment dose being taken. Alternatively the combination of agents may have resulted in too great a reduction in airway mucus viscosity so that cilia were unable to function effectively (the mucociliary escalator would not clear pure water for example).
Given the potential problems of tolerability with mannitol in our children, recruiting subjects to future studies may depend on altering certain aspects of our study design. It may be appropriate to recruit children with milder chest disease (particularly if mannitol is seen as potentially beneficial early in the disease process). A lower dose of mannitol may improve tolerability. It would be of interest to include newer outcome measures in any future therapeutic trial. The mechanism for the apparent mutual antagonism of mannitol and rhDNase combined also requires further study. The effect of mannitol on airway inflammation and sputum rheology remains uncertain and is an interesting area for future research.
In summary, we have shown in the first therapeutic trial of inhaled mannitol in children with CF that it is potentially at least as effective as rhDNase. The combination of both treatments was not beneficial. There is marked variation in response to both treatments and in addition to HS, and individuals who do not benefit from either rhDNase or HS may warrant a trial of inhaled mannitol. Cough and bronchoconstriction may limit the tolerability of mannitol for some children.
Acknowledgments
The study was funded by Pharmaxis, NSW, Australia.
REFERENCES
Footnotes
Competing interests The study was sponsored by Pharmaxis, NSW, Australia.
Ethics approval Approval for this study was obtained for the Royal Free Hospital Ethics Committee, London.
Provenance and Peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves