Article Text

Abstract
The use of retinoids to induce human lung regeneration is under investigation in a number of studies in patients with chronic obstructive pulmonary disease (COPD). Retinoic acid (RA) has complex pleiotropic functions during vertebrate patterning and development and can induce regeneration in a number of different organ systems. Studies of retinoid signalling during lung development might provide a molecular basis to explain pharmacological induction of alveolar regeneration in adult models of lung disease. In this review the role of endogenous RA signalling during alveologenesis is explored and data suggesting that a number of exogenous retinoids can induce regeneration in the adult lung are discussed. Current controversies in this area are highlighted and a hypothesis of lung regeneration is put forward. Understanding the cellular and molecular mechanisms of induction of regeneration will be central for effective translation into patients with lung disease and may reveal novel insights into the pathogenesis of alveolar disease and senescence.
Statistics from Altmetric.com
The lung is the primary organ for gas exchange across mammalian species, providing an interface between blood and air in a structurally stable environment. As gas exchange is a passive process, a large thin surface area is required for efficient supply of oxygen and removal of carbon dioxide. Gas exchange occurs in alveolar saccules, alveoli and alveolar ducts. Alveolar formation (or alveologenesis) occurs predominantly during a developmental restricted early postnatal period in rats, mice and humans.1–5 The cellular and molecular regulation of this complex developmental event is not well understood. Here we discuss a growing body of evidence highlighting the role of endogenous retinoic acid (RA) signalling in the regulation of mammalian alveologenesis, and interpret findings suggesting that pharmacological dosing with RA can induce alveolar regeneration in adult animals outside the temporally restricted period of alveologenesis. As fundamental developmental mechanisms are widely conserved across species, these findings open up the possibility of human lung regeneration as a potential therapeutic approach for patients with lung disease.
The gas exchanging tissues of the pulmonary alveoli are lined with epithelial cells (type I and type II cells) which are in direct contact with air and therefore vulnerable to injury following the inhalation of toxins, infections or reactive oxygen species. The remarkable ability of the lung to maintain function through the life of the organism must, in part, reflect the ability of epithelial, interstitial and endothelial cell populations—together with extracellular matrix—to undergo renewal, repair or regeneration over the lifetime of the animal. In other words, the gas exchanging regions of the lung must have a degree of plasticity. That this plasticity exists has been demonstrated, at least in rodents, by a body of work by Massaro and colleagues and others in a number of different experimental paradigms including re-feeding after calorie restriction6 and oestrogen replacement in oestrogen-deficient mice,7 and is exemplified by experiments showing that exogenous all-trans retinoic acid (atRA) can induce alveolar regeneration in adult rats with elastase-induced emphysema,8 partially rescue emphysema in the tight-skin mouse mutant9 and rescue dexamethasone (Dex)-impaired alveologenesis in adult mouse lung.10–12
The ability to pharmacologically induce alveolar regeneration is a novel and attractive therapeutic approach for patients with too few alveoli and a reduced gas exchanging surface area and might be applied to infants with developmental lung arrest, adults with emphysema or adults with age-related loss of alveoli.13 The restoration of functional gas exchanging tissue in humans with alveolar insufficiency would be a significant step forward in the treatment of respiratory diseases that have major public health implications where little in the way of disease-modifying therapy exists.14 Whether it is possible to induce regeneration in the human lung is not yet known. Currently, specific retinoic acid receptor (RAR) γ agonists are in phase II clinical trials in adults with emphysema secondary to α1-antitrypsin deficiency and in patients with smoking-related emphysema.
In this review we discuss the potential mechanisms by which atRA can induce alveolar regeneration in animal models by examining data highlighting the role of endogenous RA signalling that occurs during normal alveolar formation. Deciphering the complex cellular, molecular and genetic signals that regulate development and regeneration provides not only an insight into the pathogenesis of diseases where homeostatic repair processes are disrupted, but should also identify rational targets for the development of future regenerative therapies. In other biological systems, regeneration—at least in part—recapitulates development so, to understand lung regeneration, we must first consider lung development.
LUNG DEVELOPMENT
Until recently, most studies of mammalian lung development have been performed in rats,2 3 15 but the recent explosion in mouse genetics offers a highly manipulated genome which allows specific questions to be asked about the role of candidate developmental genes. These studies, coupled with lineage tracing experiments, should enable progress to be made in deciphering the complex patterning mechanisms that underpin lung development. Current knowledge of these mechanisms is, at best, sketchy. Normal function of the lung requires the correct temporal and spatial sequence of developmental mechanisms. Understanding how these fundamental signalling pathways that appear to be active—rather like switching lights on and off at different times in different places—are regulated and interact will be central to understanding not only mechanisms of alveolar development, but also alveolar disease and senescence.
The lung arises from foregut endoderm during early development of the embryo. Paracrine signalling between the extending epithelium and surrounding mesenchyme is involved in patterning the developing lung bud.16 Lung development proceeds through four morphologically distinct but continuous embryonic phases that include branching morphogenesis (embryonic day, E9–E12 in mice), the pseudoglandular stage (E12–E15 in mice), canalicular stage (E15–E17 in mice) and saccular stages (E17–birth in mice). In rats and mice, pups are born with no true alveoli but thick-walled alveolar saccules and alveolar formation occurs entirely as a postnatal event.1–3 Human alveologenesis occurs from around 24 weeks, the current limit of premature survival, the majority of human alveolar formation occurring during the early postnatal period, certainly up to 18 months5 and possibly into later childhood even early adulthood.
A DISTAL LUNG SIGNALLING CENTRE AND INDUCTION OF ALVEOLOGENESIS
Alveoli are generated through a number of different possible mechanisms that are differentially regulated, the best characterised being alveolar septation where alveolar saccules are subdivided by the eruption of a secondary crest from the primary saccular wall. Septation is a developmentally regulated process that occurs in mice from postnatal day 4 (P4)–P14 (fig 1). The relative timing of alveolar septation is species-dependent and has been proposed to be regulated by oxygen requirements at birth.17 Morphological changes driven by proliferation of type II epithelial cells and fibroblasts together with differentiation of type I epithelial cells, transformation of a double capillary system into a single complex capillary network, and apoptosis of specific cell populations resulting in thinning of the alveolar wall and increased architectural complexity characteristic of adult lung tissue.

Increasing alveolar complexity during postnatal alveolar formation in the mouse lung. High power images of the developing postnatal mouse lung. All images are taken at the same magnification. (A) The smooth walls of the large alveolar saccules are clearly visible in the P1 mouse lung. (B) With subsequent alveolar development to P4, the saccules become subdivided by the eruption of thick-walled primary alveolar septa (arrows). (C) At P9, numerous secondary septa erupt from the primary septal wall (arrowheads). (D) At P15, the alveoli have thinner alveolar walls and resemble the structure of the adult lung (E). Scale bar 50 µm. P, postnatal day.
The molecular signals that induce septal eruption are not yet known, but retinoid signalling in pulmonary lipofibroblasts appears to play a key role.18 These cells resemble stellate cells of the liver and are the major retinoid storage cell of the lung.19 Lipofibroblasts contain many components of the retinoid signalling pathway including receptors and binding proteins20 and can synthesise biologically active RA.21 Elastin is a major component of the extracellular matrix with low turnover in steady state and contributes to the elastic properties of the lung. Lipofibroblasts respond in an autocrine manner to RA by upregulation of elastin gene transcription.22 Lipofibroblasts are found in septating tissue adjacent to alveolar type II epithelial cells.2 In vitro studies demonstrate that RA can induce proliferation in immortalised type II cells in vitro, suggesting a paracrine action of RA on epithelial type II cells during alveologenesis23 and a paracrine manner on endothelial cells to induce gene transcription.21 Lipofibroblasts are progenitors of myofibroblasts, a subset of fibroblasts that have contractile components that are required for alveolar septation to proceed,24 perhaps by facilitating septal elongation. These findings suggest that lipofibroblasts are ideally placed to establish a local morphogenetic signalling centre to coordinate alveolar septal eruption.
Interestingly, morphometric studies suggest that septation alone does not account for the entire increase in alveolar tissue that occurs in the postnatal period, and that alveoli might be generated by the transformation of distal bronchioles into alveoli, called retrograde alveolisation.25 Recently, a subpopulation of cells known as bronchoalveolar stem cells (BASCs) has been identified that are located at the interface between the terminal bronchiole and the alveolar epithelium—the bronchioalveolar duct junction. These cells co-express Clara cell markers (CC10) typical of distal bronchiolar epithelium and type II cell markers (SP-C) seen in the alveolar epithelium. Isolated BASC populations in vitro have been shown to self-renew—a requirement for a stem cell population—and can differentiate into cells that express markers characteristic of bronchial epithelium, alveolar type I cells or alveolar type II cells.26 The role of these cells in alveolar formation has not yet been defined, but the location and multilineage potential of this population suggests that, under the right developmental cues, these cells might contribute to alveologenesis.
There is some evidence that matrix turnover is higher in subpleural regions of the lung outside the period of septation, and this is accompanied by a shift in the location of lipofibroblasts from central regions during septation to the periphery after septation ceases.27 In studies of alveolar wall cell proliferation during murine alveologenesis, two discrete patterns of proliferating cells have been identified: a central pattern that is temporally associated with septation and a peripheral subpleural pattern that extends into adulthood.28
ENDOGENOUS RETINOIDS AND RA SYNTHESIS IN THE POSTNATAL MOUSE LUNG
Retinoids are the biologically active metabolites of vitamin A (retinol) and have a central role in a number of developing and regenerating biological systems including developing and regenerating limb, skin and central nervous system.29 In these quite different models, common themes emerge to explain exogenous effects of retinoids by the endogenous effects of RA during development.30
Mammals are unable to synthesise retinoids de novo and are dependent on dietary sources either in the form of β-carotenes from plants or retinyl esters from animal sources. Following absorption from the gut, retinoids are stored in a number of organs prior to utilisation. The lung is second only to the liver as the major retinoid storage organ. The requirement of retinoids for the integrity of epithelial tissues, including the lung, has been known about for many years.31 32 Adult lung contains a wide range of different retinoids including all-trans RA (atRA), 13-cis RA, 14,4-di-dehydroretro RA, 4-oxoRA, anhydroretinol and retinyl esters.28 The availability of active RA is modulated by a number of factors including the distribution and activity of RA synthesising enzymes and RA modulating enzymes including cytochrome P450 enzymes such as CYP26 1a1 and CYP26 1b1. The retinaldehyde dehydrogenase enzymes (RALDHs) are the rate-limiting step in local RA production, and their spatial distribution in the embryo usually defines RA-dependent regions or periods of development.29
In addition to endogenous retinoids, lung tissue contains RALDH enzymes that are temporally and spatially regulated during alveologenesis in the mouse (RALDH-1 and RALDH-2). This study suggests the expression of different RALDH enzymes are associated with different patterns of alveolar wall cell proliferation; RALDH-1 is associated with the tightly regulated central pattern corresponding to the period of alveolar septation and RALDH-2 with a peripheral subpleural pattern supportive of the circumferential addition of alveoli. RALDH-1 is located in bronchial epithelial cells and in cells within the alveolar wall but not pleural tissue. RALDH-2 is identified in bronchial epithelial tissue and pleura but not in the alveolar septum.28
RETINOID BINDING PROTEINS DURING MURINE ALVEOLOGENESIS
Both retinol and RA are bound in the cytoplasm by binding proteins—cellular retinol binding protein (CRBP-I and -II) and cellular retinoic acid binding protein (CRABP-I and -II). These families of cytoplasmic binding proteins regulate the biological action of retinol and RA, modulating both the availability of free retinoid and ensuring specificity between the retinoid and its metabolic enzymes (fig 2).33 Previous Northern blot analysis of CRBP-I and CRABP-I mRNA has demonstrated upregulation of both genes during alveologenesis in both whole rat lung tissue and isolated lipid-laden fibroblasts.20 Dexamethasone administered to postnatal rat pups between P4 and P14 disrupts alveolar septation34 and results in downregulation of CRBP-I and CRABP-II.35 Retinoid binding proteins are located in parenchymal lung tissue during alveologenesis.36 The temporal rise in the CRBP-I and CRABP-I towards the end of alveolar septation in both rats and mice suggests that these proteins might regulate the availability of free retinol and RA to end septation.
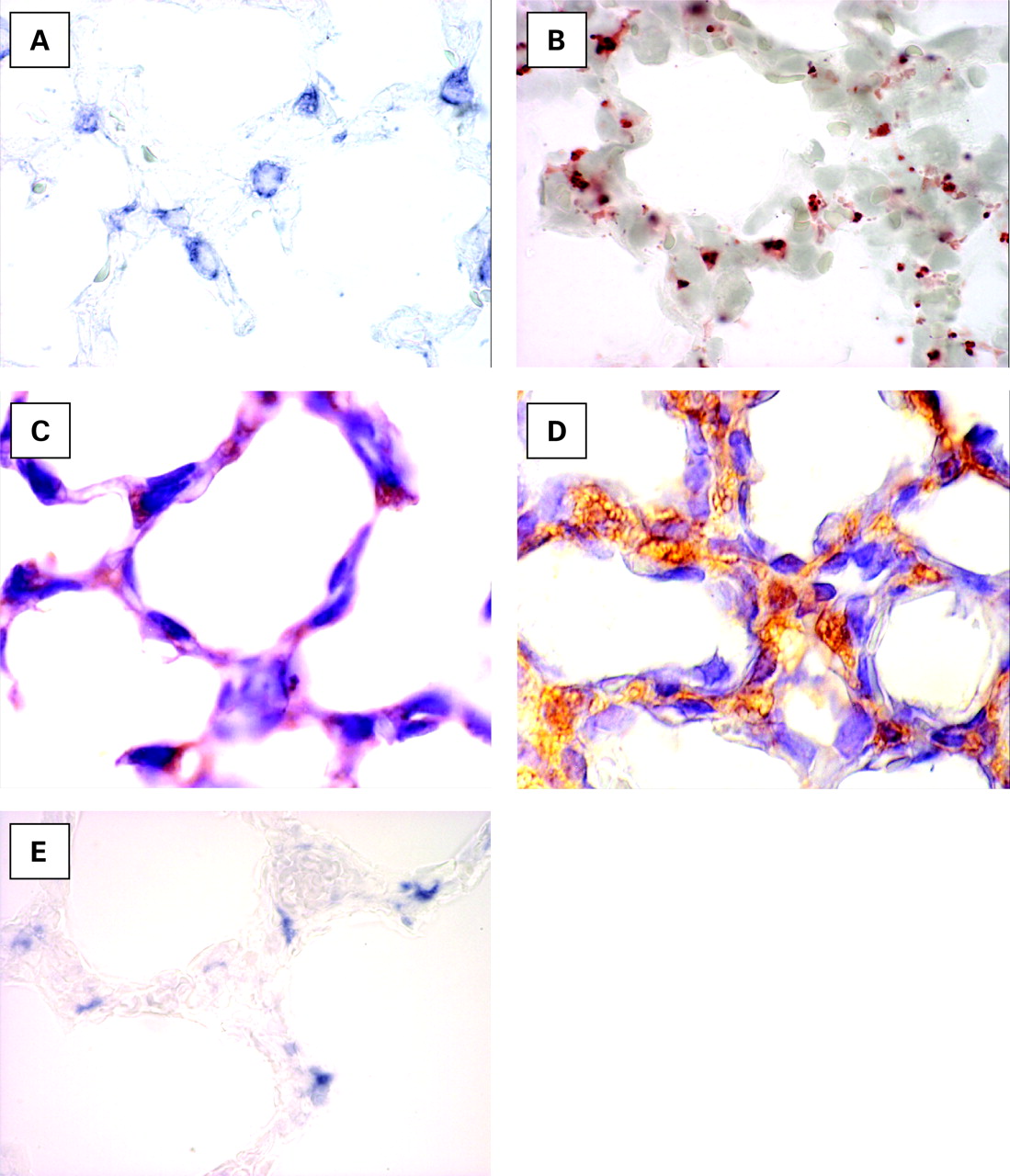
Type II cells, lipofibroblasts, retinoic acid (RA) signalling proteins and elastin mRNA expression during murine alveologenesis. Cell types and RA signalling proteins associated with alveologenesis. (A) In situ hybridisation with digoxigenin-labelled antisense riboprobes to surfactant protein C mRNA (blue colour) restricted to type II pneumocytes. These cells proliferate in response to RA in vitro. (B) Oil red-O highlights the presence of lipid droplets (red stain) in retinoid-containing lipofibroblasts on frozen sections of alveolar parenchyma. (C) Immunohistochemistry demonstrating retinaldehyde dehydrogenase-1 (RALDH-1) protein (brown staining), an endogenous RA-synthesising enzyme in specific alveolar parenchymal cells. (D) Cellular retinol binding protein (CRBP-1) immunohistochemistry (brown stain) reveals widespread alveolar parenchymal distribution of the retinoid binding proteins. (E) In situ hybridisation with digoxigenin-labelled antisense riboprobes (blue colour) to tropoelastin demonstrating asymmetrical tropoelastin mRNA expression, an RA regulated target gene, in the presumptive erupting alveolar septa.
RETINOIC ACID RECEPTORS DURING MURINE ALVEOLOGENESIS
RA exerts its effects via nuclear receptors, retinoic acid receptors (RARs) which act as ligand-activated transcription factors to alter the pattern of gene transcription of a cell. There are three subtypes of RAR (α, β and γ), each of which has a number of isoforms through differential promoter usage and splice variants. There are seven isoforms of RARα, four isoforms of RARβ and seven isoforms of RARγ. In the mouse, RARα1, RARβ2, RARβ4 and RARγ have been identified in postnatal lung tissue and expression of these receptors changes over time.36 These data are in agreement with earlier experiments identifying RARα, β and γ in lipofibroblasts isolated from rat lung.20
The function of the individual RARs on alveolar formation has been investigated in mice using knockouts of each of the three RARs. Morphometric study of RARα−/− mice found no difference between control and knockout animals at P14 (the end of alveolar septation), but at P50 both the number of alveoli and surface area is reduced compared with controls which suggests that RARα has no effect on septation but has a role on non-septation alveologenesis.37 The effect of RARβ on alveolar formation has been determined by analysis of gene deletions and by use of specific RARβ agonists38 which show that RARβ−/− animals septate earlier and at a faster rate than controls, and that dosing with RARβ-specific agonists impairs septation. These data suggest that RARβ is a negative regulator of septation although, interestingly, analysis of a different RARβ mutant gave different results with a reduction in the gas exchanging surface area per lung volume.39 Deletion of one RARγ allele has been found in the compound RXR/RARγ+/− mice to result in a reduction in whole lung elastic tissue, number of alveoli and an increase in the distance between alveolar walls and mean chord length (Lm). In addition, elastin transcription in lipofibroblasts was reduced at P14.40 These data suggest that RARγ is a positive regulator of septation.
RETINOID SIGNALLING IN HUMAN ALVEOLOGENESIS
From the data above, numerous lines of evidence point to a central role of RA signalling in alveolar formation in rodents. Is there any evidence that this developmental mechanism is conserved in man? Chronic lung disease of prematurity or bronchopulmonary dysplasia (BPD) describes a clinical syndrome of lung dysfunction characterised by a developmental lung arrest.41 The aetiology of BPD is multifactorial with prematurity, infection, mechanical ventilation, oxygen toxicity and the use of glucocorticoids all implicated. Low serum vitamin A (retinol) levels have been associated with BPD.42 Intermittent three times weekly dosing with retinol is one of the few interventions found to have a small but significant effect on reducing the incidence of BPD in extremely low birthweight infants,43 although the mechanism of this effect is not known. Interestingly, the first human mutation of RA signalling (STRA6), the recently discovered membrane receptor for uptake of circulating retinol,44 has been described in two families following a screen of infants with anophthalmos. These infants had a number of specific developmental malformations including lung hypoplasia, suggesting a requirement for correct RA signalling in human alveolar development.45
These findings suggest an evolutionary conserved pathway in which developmentally regulated endogenous RA is synthesised from stored retinoids in lipofibroblasts to establish a distal lung signalling centre. This epithelial-mesenchymal signalling centre acts in an autocrine manner on lipofibroblasts with paracrine actions on type II cells, endothelial cells and myofibroblasts to initiate—and perhaps coordinate—alveolar septal eruption and regulate alveologenesis. This model provides a molecular framework to consider the effects of exogenous RA on the induction of regeneration.
To determine whether the Dex effect also occurs in the mouse, neonatal mouse pups were dosed from P2–P14 with Dex or dilutent and allowed to develop into adults with no further intervention. This generates adult mice in which alveolar development has been disrupted and spontaneous alveolar regeneration does not occur (at least until P150).52 To investigate the physiology of this model we used spontaneously breathing, whole body plethysmography under conditions of physiological stress to demonstrate that neonatal Dex dosing results in increased tidal volume, minute volume and a reduced rate in adult animals (ie, Dex-dosed animals breathe more slowly, for longer and work harder; Stinchcombe et al, unpublished). The structural and functional characteristics of this model of disrupted development share many characteristics with human BPD/COPD.
Using this Dex model it has been shown that 10 doses of RA from P40–P52 results in changes in morphology and morphometry suggestive of successful alveolar regeneration (a reduction in Lm and increase in surface area to that of control animals) when assayed at P90.10 Using this model of airspace enlargement, other molecules can be assayed for potential regenerative effects. We have tested eight other retinoids including 13-cis RA, 9-cis RA, retinol, 4-oxo RA, a pan RXR agonist and RARα, RARβ and RARγ agonists in our murine Dex model, some of which induce regeneration (RARα, RARγ, 4-oxo RA) but others do not (13-cis RA, retinol, pan-RXR agonist) (fig 3).11 Using the Dex/RA protocol and manipulating the genetic background of the mouse, we can determine which downstream genes are required for regeneration to occur and which are redundant. For example, RA can induce regeneration following the Dex/RA protocol on an RARβ null mutant background, suggesting that RARβ is not required for the regenerative effect. This finding is confirmed by data demonstrating that RARβ-specific agonists do not induce regeneration in wild-type mice.11 This combination of morphometry, genetics and experimental intervention provides a potential approach for analysis of microarray-identified downstream regeneration genes.

{kind=link}
{kind=link}
{kind=link}
Induction of regeneration using different retinoids. Restoration of gas exchanging surface area (SA) normalised to body weight in the dexamethasone (Dex) mouse model of alveolar insufficiency: 4-oxo RA, atRA, RARγ agonist, RARα agonist and 9-cisRA restore lost SA but retinol, pan-RXR agonists, RARβ agonists and 13-cis RA do not.
RA-INDUCED ALVEOLAR REGENERATION: CURRENT CONTROVERSIES
Given the widespread potential implications of lung regeneration, numerous studies have tried to replicate the original report by Massaro and Massaro8 in different animal models of disease. To date, seven have succeeded10 11 38 53–55 and in two other experimental paradigms—pneumonectomy and oxygen damage—atRA stimulated proliferation or protected from damage.56 57 However, a similar number of studies have failed to show any beneficial effects of atRA.58–64 The reason for these differences are not yet clear.
To investigate why some of the published studies using mice failed to identify a regenerative effect, we investigated the effect of background strain on regeneration. Our original studies were performed in the outbred TO strain but, in these experiments, we examined inbred NIHS and ICR strains.65 Using the same protocol as described above, NIHS mice gave similar results to the TOs (ie, a mean decrease in Lm in Dex-treated animals to atRA-treated animals which represents a 56% recovery towards the control Lm value). The same distribution of animals in the atRA-treated group was observed with some showing complete recovery, the majority showing a partial response and some showing no response at all. However, using this protocol, the ICR strain revealed that atRA had absolutely no effect on alveolar structure or morphometry. When the above experiment was repeated on the ICR strain but with a five times higher administered dose of atRA (10 mg/kg), successful regeneration was induced. These data suggest that RA-induced regeneration is not a strain-specific phenomenon but that there are important differences in RA pharmacokinetics between strains.
The second obvious clinical scenario where RA might have therapeutic implications is in adults with emphysema. Epidemiological data suggest that there is an inverse relationship between serum vitamin A levels and lung function in smokers with COPD.68 The NIH-funded FORTE study (Feasibility of Retinoids for the Treatment of Emphysema) investigated dosing with atRA 1 mg/kg, 2 mg/kg or 13-cis RA in a 6-month trial with a 3-month crossover period and subsequent 18-month observation period.69 Retinoids were generally well tolerated in this patient group with only minor side effects (commonly rash, hypertriglyceridaemia and mild alterations in liver function tests). There were no significant improvements in the CT densitometry score, gas transfer or quality of life score at the end of this feasibility study, but there was evidence of some biological activity, with a change in carbon monoxide transfer factor (initial decline with later recovery) in patients who received the highest doses of RA with high plasma levels. The significance of this finding is as yet unclear. atRA can induce pulmonary toxicity as an acute retinoic acid syndrome. Unfortunately, the crossover design of the FORTE trial resulted in no true placebo group for long-term follow-up. The reported change in carbon monoxide transfer factor could therefore represent either drug-induced toxicity or an alveolar remodelling process.
Specific RARγ selective agonists are currently in longer Pharma-sponsored placebo controlled phase II clinical trials in patients with emphysema secondary to α1-antitrypsin deficiency and in patients with moderately severe COPD due to smoking. These molecules are at least as potent as atRA at inducing alveolar regeneration in the Dex mouse model, do not induce their own metabolism and have more predictable pharmacokinetics so that higher drug levels can be maintained. The outcome of these trials is expected in the next few years. These studies highlight the usefulness of appropriate animal models in the rational design of candidate regenerative therapies for the treatment of human lung disease.
The induction of endogenous repair or regenerative pathways using exogenous agents represents a promising and exciting therapeutic approach for patients with lung disease. The development of a mouse model allows the cellular and molecular biology of the induction of alveolar regeneration to be studied in detail. Translational studies with the goal of inducing lung regeneration are now underway in patients with emphysema. Just 10–15 years ago the idea that the adult lung had the capacity to undergo regeneration was viewed with scepticism. The fact that human trials are now underway is remarkable. The next 10–15 years’ research promises huge advances in this field.
REFERENCES
Footnotes
Funding: This work was supported by a Wellcome Training Fellowship (MH), a Wellcome Trust project grant (MM and MH), Wellcome VIP Award (ÅG) and an MRC Clinical Fellowship (SS).
Competing interests: None.