Article Text

Abstract
Background: The pathophysiological basis of severe respiratory syncytial virus (RSV) bronchiolitis in infancy is poorly understood and has hindered vaccine development. Studies implicate the cell-mediated immune response in the pathogenesis of the disease. A recent twin study estimated a heritable contribution of 22% to RSV bronchiolitis. Genetic epidemiology provides a new approach to identifying important immune determinants of disease severity.
Methods: A comprehensive high-density gene-region association study for severe RSV bronchiolitis in infancy at 5q31 across 11 genes including the Th2-cytokine cluster was performed. A haplotype tagging approach was used to analyse genetic variation at 113 single nucleotide polymorphisms (SNPs) in 780 independent cases and 1045 controls. The study had sufficient power to detect small effects, perform extensive haplotype analysis and analyse both a principal phenotype and a refined age-limited phenotype enriched for first-exposure RSV infection.
Results: SNP associations were found at IL4 and a highly significant risk haplotype was identified across IL13 CNS-1 and IL4 (odds ratio 1.69, p<0.0001), present in both case-control and family-based analyses. All associations were strongest for a phenotype limited to <6 months of age, implicating this locus in primary RSV disease. The same risk haplotype has previously been shown to be associated with increased IL13 expression.
Conclusions: A haplotype at IL13–1L4, which is associated with increased IL13 production, confers an increased risk of severe primary RSV bronchiolitis in early infancy. This study, together with previous studies implicating the same locus in atopic sensitisation, suggests that primary RSV bronchiolitis and atopy share a genetic contribution at the IL13–IL4 locus.
Statistics from Altmetric.com
Respiratory syncytial virus (RSV) bronchiolitis is responsible for between 18 000–75 000 hospital admissions and 200–500 deaths annually in the USA.1 The pathophysiological basis of severe RSV bronchiolitis in infancy is poorly understood and this has hindered vaccine development. Studies consistently implicate the host response as an important determinant of disease severity, and many immunological studies have linked the Th1/Th2 spectrum of T cell differentiation in disease pathogenesis.2–4
The possibility of a genetic vulnerability is supported by a recent twin study comparing monozygotic with dizygotic twins, which showed RSV bronchiolitis in infancy to have a heritable contribution of 22%.5
Genetic epidemiology provides a new approach to dissecting the pathogenesis of complex disease traits. Study design is paramount, and many early studies have been difficult to replicate because of small sample size, consequent lack of power, phenotypic heterogeneity between studies and failure to accommodate for population substructure or multiple testing. Confidence in results can be gained through replication in independent studies.6
Animal studies of RSV bronchiolitis have been used extensively to investigate the immune response to RSV infection.7 8 Less is known about the immune response of affected infants. Genetic epidemiology represents a powerful approach to identifying important immune determinants of severity in human disease.
Three recent genetic association studies of RSV bronchiolitis have identified associations for promoter polymorphisms in the Th2 cytokine IL4.9–11 These studies were performed in small cohorts with limited power and sampled a small number of candidate polymorphisms only. Moderate associations were reported without accommodation for multiple testing.
In this study we performed a high-density comprehensive gene-region association study for severe RSV bronchiolitis in infancy at 5q31, a region containing the Th2 cytokine cluster and many other important immune genes. We applied a haplotype tagging approach to sample variation at 113 single nucleotide polymorphisms (SNPs) across the genes IL3, GMCSF, P4HA2, RIL, SLC22A4, SLC22A5, IRF-1, IL5, RAD50, IL13 and IL4, and across all known intergenic regulatory elements. We used 780 independent cases and 1045 population controls of documented white European ancestry, with sufficient power to detect small effects, perform extensive haplotype analysis and accommodate for multiple testing. Our large sample size gives us sufficient power to concentrate specifically on first exposure RSV in the immunologically naïve infant by performing analysis on an age-limited phenotype (<6 months of age). This enabled us to dissect out the phenotype of primary RSV disease form RSV-induced wheeze or atopic wheeze precipitated by RSV infection that may present towards the end of the first year of life. We identified a highly significant risk haplotype across IL13 CNS-1 and IL4 which we have previously shown to be associated with increased IL13 expression.12
METHODS
DNA samples and DNA extraction
The Oxford RSV DNA Archive has been described elsewhere.13 In this study, 782 cases, 1045 cord blood controls and 673 families were used in association analysis.
Identification and selection of SNPs across 5q31
The public databases CHIP Bioinformatics14 and Project Ensembl were searched for all known SNPs across 656 kB of the 5q31 gene region. PubMed was searched for relevant literature relating to SNP discovery, disease association and functional molecular genetics at 5q31.
Strategy in deciding which SNPs to genotype was directed by two aims: to comprehensively describe the haplotype diversity of the 5q31 gene region and to enrich for SNPs with potential functionality.
Under the hypothesis of evolutionary constraint, cross species conserved non-coding sequences (CNS) may contain functional DNA important in gene regulation. SNPs in CNS regions were identified by human-mouse sequence homology studies using the web-based program VISTA.15
Transcription factor (TF) binding sites were identified using the program Matinspector. SNPs predicted to disrupt TF sequence motifs were again considered as important candidates for association analysis.16
In selecting SNPs for genotyping, priority was given to those SNPs previously validated in a white European population with a predicted frequency of >5%, to those positioned within a gene, located in particular within the promoter, exons or within 300 base pairs of an intron/exon boundary, to those SNPs in or within 300 base pairs of a CNS, and to those SNPs at a potential TF binding site. All SNPs with a previous positive disease association in the literature were also included.
Case definition, study phenotypes and control samples
Cases
The Oxford RSV archive contains in excess of 1200 cases. For the current study, the following phenotypes were defined and studied.
The principal phenotype is defined by the following criteria: a clinical diagnosis of bronchiolitis (breathlessness, chest wall recession and inspiratory crepitations on auscultation), evidence of RSV on nasopharyngeal aspirate, requirement for oxygen and/or nasogastric feeds during hospital admission, age <1 year at time of hospital admission. Only children with two white European parents were included in the study to avoid the effects of population substructure.17 This phenotype is largely consistent with previous studies of RSV disease in infancy at the IL4 locus, although these studies enrolled children up to 2 years of age.
The second phenotype used in this study is a subgroup—here called the refined phenotype—which, in addition to the above criteria, also excludes those children with known risk factors for RSV disease (prematurity, chronic lung disease, congenital heart disease) and is restricted to infants under 6 months of age at the time of illness. This phenotype enables us to study more clearly the impact of polymorphism at 5q31 in the immunologically naïve infant during primary RSV exposure. Demographic data on both phenotypes are shown in table 1.
Controls
The 1045 population controls used in the study were sequential cord blood samples taken from white European infants born at the John Radcliffe Hospital, Oxford. Population controls can be used in association studies where the disease phenotype is comparatively rare in the general population. This is the favoured approach taken by the Wellcome Trust Case Control Consortium GWAS initiative18 and is appropriate for severe RSV bronchiolits which has a frequency of 1–3% in the general population.
Haplotype tagging approach to association study
A haplotype tagging approach to association was implemented. In this approach, many SNPs are selected and genotyped in a small representative group of population samples and the haplotypic relationships between these SNPs is inferred. A subset of haplotype tagging SNPs (htSNPs) which together comprehensively describe all the genetic diversity of the region can then be selected and taken forward to genotyping in the cases and controls. This reduces genotyping costs when using large cohorts.
In this study, all selected SNPs were first genotyped in 32 representative white European family trios and common population-specific haplotypes were inferred using the algorithms Phamily and PHASE.19 The pattern of linkage disequilibrium across the region was analysed using the programs HaploXT20 and MARKER,21 and revealed well demarcated haplotype blocks. An efficient subset of htSNPs was selected within each block independently, using methods described elsewhere.22 A total of 48 htSNPs were selected and taken forward for genotyping in the case-control association study.
Study design
In total, 782 cases and 1045 controls were genotyped in this association study.
As a cost-saving strategy, all 48 htSNPS were first genotyped in 420 cases and 576 controls (cohort 1). htSNPs with the strongest positive associations in cohort 1, together with SNPs required to generate haplotypes across IL4_IL13, were then genotyped in the remaining 362 cases and 469 controls (cohort 2).
The primary analysis for this study relates to the seven SNPs genotyped in the total cohort (amounting to a total of 782 cases and 1045 controls). These were used in single SNP and haplotype analysis. The data for the 48 SNPs genotyped in cohort 1 were also analysed independently in an extensive haplotype analysis spanning 5q31.
The seven htSNPs genotyped in the total cohort were also tested in a family-based association analysis in 673 of the cases where DNA from both parents was available.
Genotyping and data curation
Methods of genotyping and curation have been described elsewhere.23 Briefly, all genotyping was performed using Sequenom MassArray using whole genome preamplified DNA. Primers and multiplexes were designed using SpectroDESIGNER. Curation of genotyping calls was implemented using SpectroTYPER. SNP assays were accepted if SNP frequency exceeded 5%, the assay was in Hardy-Weinberg equilibrium in controls (HWE χ2 p>0.05) and genotyping success exceeded 80%.
Statistical analysis
Single SNP analysis
Only SNPs with a population frequency of >5% were included in the analysis. For each SNP, allelic association statistics were generated using a 2×2 χ2 test (1df) with the odds ratio calculated for minor allele versus major allele. Genotype association statistics were generated using a 2×3 χ2 test (2df).
Haplotype analysis
Phase 2.1.1 implementing haplotype probability assignments was used to generate haplotypes.19 Each individual may be allocated several different haplotype pairs with different probabilities and these are incorporated into the association analysis such that a single individual may contribute to the overall frequency of several different haplotype pairs.
Similar haplotypes that differ at one or a small number of sites are likely to be related by genealogy and, by virtue of their shared ancestry, these haplotypes are likely to share other unidentified recent alleles. Grouping these haplotypes together by genealogy will therefore theoretically increase the power to detect unobserved variants in association analysis. This forms the basis of cladistic haplotype association analysis.
Haplotype clades within each haplotype block were generated by hierarchical clustering using the program NEIGHBOUR. All within-block haplotype analysis was performed using 2×2 χ2 tests generated for each clade versus all other clades.
Family-based association statistics were calculated using the transmission disequilibrium test (TDT).24
RESULTS
Population haplotypes and selection of htSNPs
Using the criteria outlined above, 152 SNPs of interest were selected for genotyping in population samples; 113 SNPs satisfied Hardy-Weinberg equilibrium, had a genotyping success rate exceeding 80% (median 96.9%, mean 94.8%) and a population frequency of >5%. Supplementary figure 1 published online only shows the distribution of SNPs identified and genotyped. Information including genotyping data for the 113 successful SNP assays can be found in supplementary table 1 in the online supplement. Genotyping data for the 113 SNPs were used to generate long-range population haplotypes across the gene region. Patterns of pairwise LD across the region were analysed using the metric r2 and demonstrate seven clearly demarcated juxtaposed haplotype blocks spanning the region (fig 1). Haplotype clades generated using hierarchical clustering revealed between three and five common haplotype motifs within each haplotype block. This small number of haplotype motifs describes between 75% and 94% of all observed haplotypes within each haplotype block. A total of 48 htSNPs were selected for genotyping in case-control 1. Pairwise r2 statistics, haplotype blocks, haplotype clades and selected htSNPs are shown in fig 1.

The 5q31 gene region used in association analysis. Annotations include pairwise r2 statistics >0.3 for 113 single nucleotide polymorphisms (SNPs) across 11 genes, haplotype blocks, haplotype clades identified using hierarchical clustering and selected haplotype tagging SNPs (htSNPs) for case-controls 1 and 2.
Case-control study
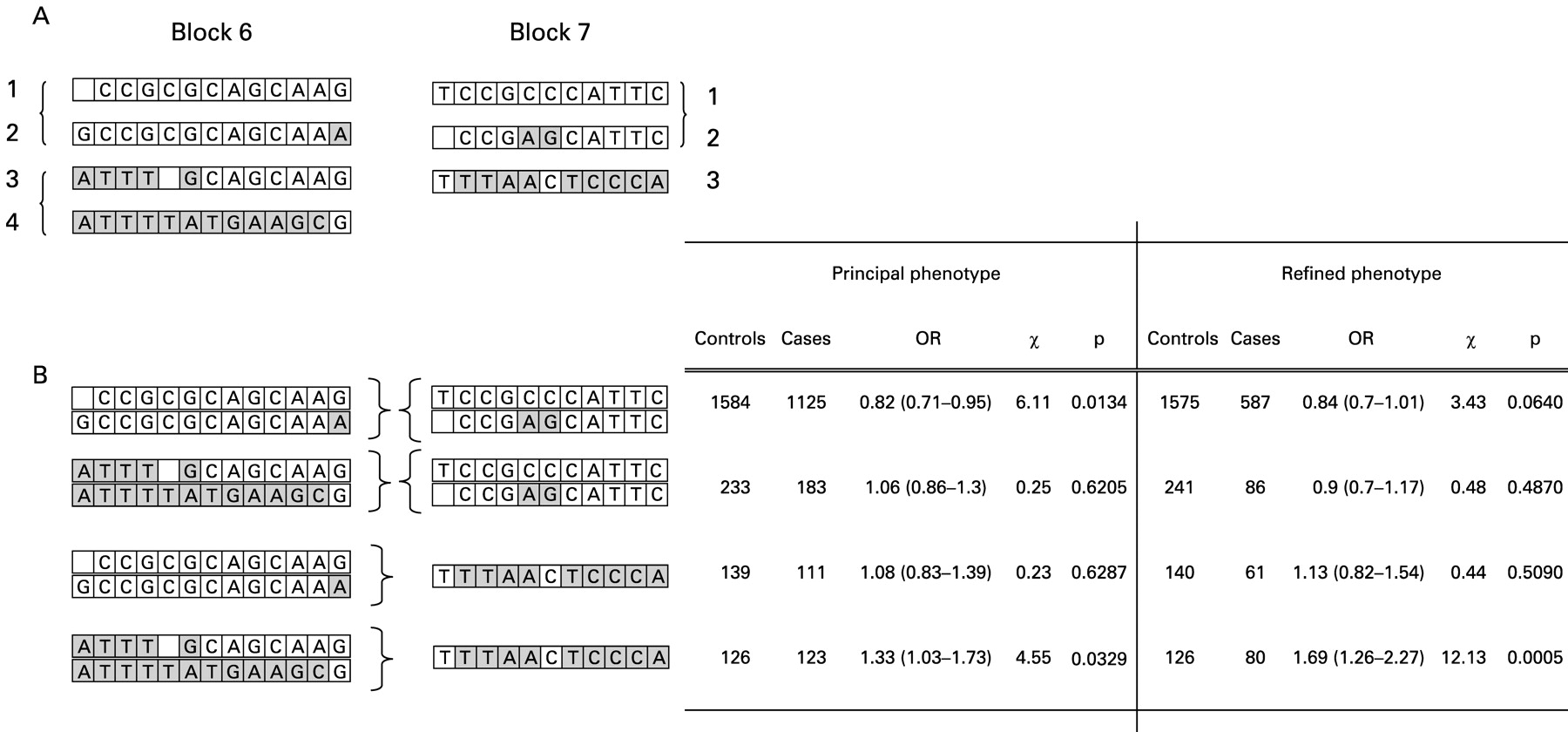
Forty-eight htSNPs were genotyped in 420 cases and 576 controls of documented white European ancestry in cohort 1. Genotyping success and association statistics for all 48 htSNPs for both principal and refined phenotypes are shown in supplementary table 2 available online. SNPs with positive associations in cohort 1 or those used later in haplotype analysis are shown in tables 2A and 3A for the principal and refined phenotypes, respectively. In the analysis of the principal phenotype, significant association results are apparent for one SNP in GMCSF, two SNPs in SLC22A5 and five SNPs in IL4. In the analysis of the refined phenotype, the associations at GMCSF and SLC22A5 are no longer present. However, the IL4 association is stronger and apparent for seven SNPs spanning CNS-1, the IL4 promoter and IL4 coding region. The strongest effect is seen for the IL4 promoter SNP rs2243250 (genotype: 2×3 χ2, p<0.003; allele: OR 1.64; 2×2 χ2, p = 0.0024). Cladistic haplotype association analysis in all seven haplotype blocks for cohort 1 produced association results in keeping with single SNP statistics but revealed no stronger effects. These are shown in supplementary table 3 available online.
Seven SNPs from cohort 1 were genotyped in a further 362 cases and 469 controls in cohort 2. These seven SNPs were selected either because they had the strongest SNP associations at IL4 and SLC22A5 in cohort 1 or were necessary to generate haplotypes in the vicinity of IL4–IL13. These seven SNPs were therefore genotyped in a total cohort of 782 cases and 1045 controls. Results for the seven SNPs genotyped in the total cohort are shown in tables 2B and 3B for the principal and refined phenotypes, respectively. A highly significant result is seen for SNPs across IL4 using the refined phenotype. Rs2243250 gives the strongest result with an odds ratio of 1.48 (p<0.001). Bonferroni correction for multiple testing is a conservative approach in the context of high LD as SNPs in this situation do not represent entirely independent tests. The results are, however, still significant when correction is made for 48 independent single locus tests (IL4 rs2243250, p = 0.03). Using methods which account for LD between tested SNPs,25 26 the effective number of independent SNPs for this dataset is calculated at 25.1, giving an overall experiment-wide significance threshold required to keep the type I error rate at 5% of p = 0.002. The results at the IL4 locus (p<0.001) clearly remain significant after accommodation for multiple testing.
Cladistic haplotype analysis for the total cohort within block 7 produced association results in keeping with single SNP statistics but revealed no stronger effects, as the minor allele for all SNPs including rs2243250 are exclusively carried by clade 3 haplotypes.
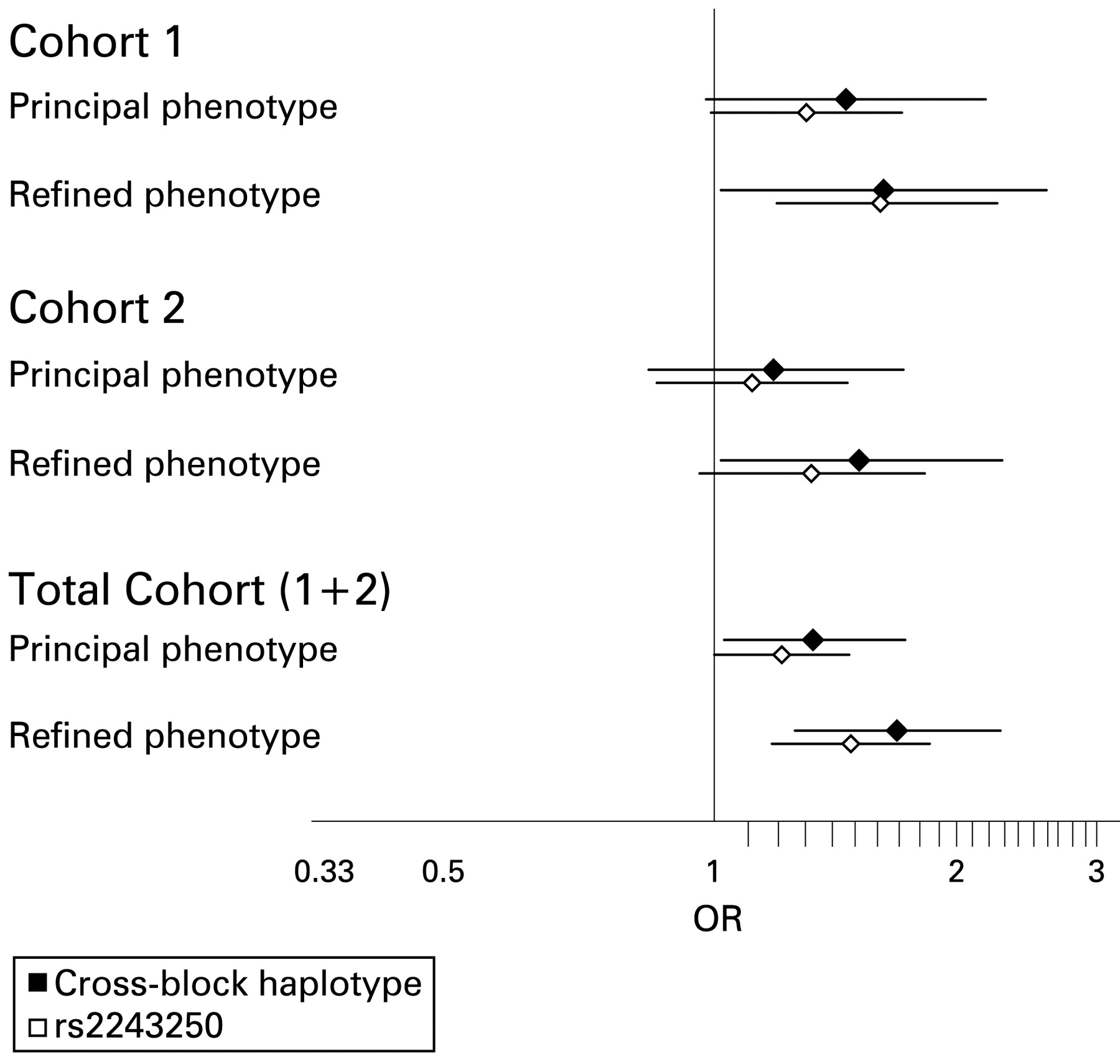
It is recognised that haplotype block boundaries defined in block analysis are not absolute, with some haplotypes crossing block boundaries intact. It is therefore difficult to delineate the source of a positive association to the haplotype block which contains the association signal. In order to address whether the haplotype clade with association seen at IL4 in block 7 extends across CNS1 and IL13, cross-block haplotypes between blocks 6 and 7 were inferred with Phase 2.1.1 using five SNPs genotyped in the total cohort. Analysis of the five SNP cross-block haplotypes is shown in fig 2. For both the principal phenotype and the refined phenotype, the haplotype association signal detected across IL4 in block 7 segregates to a specific cross-block haplotype. The association in block 7 is stronger if it is in continuum with clades 3/4 in block 6 (principal phenotype OR 1.33 (1.03–1.73), p = 0.05; refined phenotype OR 1.69 (1.26–2.27), p<0.001) and absent if it is in continuum with clades 1/2. When the same cross-block haplotypes are generated and analysed in cohort 1 and cohort 2 independently, a significant association is found independently in both cohorts for the refined phenotype and a trend to significance with the principal phenotype. In all cases, the cross-block haplotype association is stronger for the refined phenotype than for the principal phenotype and stronger than for any single SNP, suggesting the source of the association signal is an unobserved SNP for which the cross-block haplotype is a sensitive marker. The results are summarised in fig 3.

Cross-block haplotypes were generated across the region spanning IL13 CNS-1 and IL4. (A) Within-block haplotype clades for blocks 6 (IL4) and 7 (IL13 and CNS-1). (B) Combination of within-block haplotype clades across blocks. The association seen for clade 3 in block 7 is only present when in combination with clades 3/4 in block 6. This risk haplotype has a stronger association than any single SNP or within-block haplotype. OR, odds ratio.

{kind=link}
{kind=link}
{kind=link}
Summary of association statistics for cohort1 1, cohort 2 and total cohort data for the single SNP with the strongest association (rs2243250) and for the cross-block risk haplotype. In all datasets the effect is stronger for the cross-block haplotype than for the single SNP, and stronger for the refined age-limited phenotype than for the principal phenotype. The cross-block haplotype is independently significant in both cohort 1 and cohort 2. OR, odds ratio.
Family analysis
The transmission disequilibrium test (TDT) was employed in a family-based study using 673 cases from the case-control study where both parents were available. TDT statistics for the seven SNPs genotyped in families are shown for the principal phenotype and refined phenotype in table 4. The family study is less well powered than the case-control study and, although no single SNP results are significant, a trend exists for both phenotypes at the IL4 SNPs with a distortion in transmission of allele 2 at rs2243250 of 53.8% in the principal phenotype group and 55.2% (OR 1.23) in the refined phenotype group. The cross-block haplotype association seen in the case-control study was tested for the refined phenotype in the family study and showed a significant increase in transmission distortion to 59.3% (OR 1.46, p = 0.048). The magnitude of this effect is in keeping with that seen in the case-control study.
DISCUSSION
We have described a comprehensive large gene-region association study at 5q31 for a severe RSV bronchiolitis phenotype incorporating the genes IL3, GMCSF, P4HA2, RIL, SLC22A4, SLC22A5, IRF-1, IL5, RAD50, IL13 and IL4 and all known intergenic regulatory elements. We used a total of 780 independent cases and 1045 controls of documented white European ancestry with sufficient power to detect small effects, account for multiple testing, perform extensive haplotype analysis and analyse both a principal phenotype and a refined age-limited phenotype enriched for primary RSV exposure.
We defined a risk haplotype across IL13, CNS-1 and IL4. The strongest single SNP effect (rs2243250) and the risk haplotype effect was seen for both the principal phenotype and for the refined age-limited phenotype (age <6 months). The findings were consistently stronger using the refined phenotype for which the risk haplotype carries an odds ratio of 1.69 (p<0.001). This remained significant even after conservative correction for multiple testing. The risk haplotype carried a stronger signal than any single SNP tested. This suggests that the source of the association signal is an unobserved SNP for which the risk haplotype is the most sensitive marker. A more complex epistatic effect resulting from the genetic interaction of more than one functional polymorphism may also explain this haplotype association.
We performed functional analysis using allele-specific transcript quantification at IL13 in immortalised B cell lines, as described elsewhere.12 This technique quantifies the relative expression of mRNA produced by the two copies of a gene within a single individual. Using many individuals, gene expression can be linked to haplotype background. The results showed that the RSV risk haplotype identified here was associated with an inducible upregulation of IL13. This effect is not explained by IL13 promoter or exonic polymorphisms, suggesting that the risk haplotype, which spans other important regulatory regions including CNS-1 (between IL4 and IL13), may carry a more distal functional element.
Previous smaller genetic studies for RSV bronchiolitis at the IL4–IL13 have identified positive associations for the single SNP rs2243250 and for haplotypes carrying rs2243250.9–11 These studies were performed in small cohorts with limited power and moderate associations were reported without accommodation for multiple testing. Nevertheless, all positive associations for risk carried the minor allele T at rs2243250 and are consistent with our findings.
Previous studies have all defined the RSV phenotype using children under 2 years of age. RSV-positive children presenting with wheeze within the first 2 years of life may potentially have primary RSV bronchiolitis, RSV-induced wheeze or atopic wheeze precipitated by RSV infection. Refining the age phenotype is particularly important when studying the effects of the IL13–IL4 locus on RSV disease, since variants at this locus have also been associated with atopy and asthma in later life.27–29 Confounding due to over-representation of atopic infants may occur if an age-restricted phenotype is not applied. To capture a cleaner phenotype for primary RSV bronchiolitis, we have limited our main analysis to children aged <6 months (408 cases). We had the power to demonstrate that the effect at the IL4–IL13 locus is even stronger in this immunologically naïve group of young infants. This strongly supports a specific association at IL4–IL13 for primary severe RSV bronchiolitis.
How do the above findings integrate into the current understanding of the pathogenesis of severe primary RSV bronchiolitis? Many studies have implicated excess Th2/deficient Th1 immune responses in severe RSV disease.2–4 Research into immunological responses to vaccines in early infancy reveals initial responses to be generically Th2-skewed with reciprocal immaturity in the Th1 cytotoxic response.30 The rate at which the immune system matures in infancy is highly variable between individuals.31 Those with delay in immune maturation are exposed to a period of susceptibility where Th1 cytotoxic responses to infections like RSV may be suboptimal. Recent epigenetic work looking at neonatal T cells has demonstrated incomplete silencing of the IL13 locus in differentiating Th1 cells, suggesting that IL13 may play an important role in the Th2 skew observed in early life.32 The RSV risk haplotype identified here is associated with upregulation of IL13 and may therefore have its effect on RSV susceptibility by contributing to the persistence of a Th2 environment in some infants.
Responses to primary RSV bronchiolitis and to allergens in early life may both be affected by modulation of the cytokine milieu. Delay in immune maturation has been implicated in the development of atopy with formation of Th2 memory in response to antigen exposure in infancy. Genetic studies for atopy and asthma have implicated the same locus at IL13–IL4, and specifically rs2243250.27–29
The epidemiological association between RSV bronchiolitis in infancy and atopic sensitisation and asthma has not been consistently demonstrated in prospective studies.33 34 Both primary bronchiolitis and atopy are complex disease traits with numerous genetic and environmental contributions35 and, in the context of multiple variables, it is conceivable that epidemiological studies may show conflicting results even if a concrete relationship exists. Our study, together with previous studies on atopy, suggests that susceptibility to primary severe RSV bronchiolitis and atopy share a genetic contribution at the IL13–IL4 locus.
Acknowledgments
The authors thank Elham Sadighi Akha and Gaia Luoni for their help with genotyping.
REFERENCES
Supplementary materials
web only appendices 64/4/345
Files in this Data Supplement:
Footnotes
▸ Additional tables and figure are published online only at http://thorax.bmj.com/content/vol64/issue4
Funding: This work was funded by a Wellcome Trust Clinical Research Training Fellowship (JTF) and the MRC(UK) (DK).
Competing interests: None.
Ethics approval: Ethics approval for all sample collection was obtained from the Oxford Regional ethics committee and the Multi Region ethics committee.