Article Text

Abstract
Background: Non-cystic fibrosis (non-CF) bronchiectasis often starts in childhood with a significant impact on adult morbidity. Little is known about disease progression through childhood and the effect on growth and spirometry. This study reviews longitudinal lung function and growth in children with non-CF bronchiectasis.
Methods: The case notes of patients with non-CF bronchiectasis were reviewed retrospectively. Patients were included if at least three calendar years of lung function data were available. Anthropometric measurements and annual spirometry were analysed over both two and four consecutive years. Changes over time were assessed using Generalised Estimating Equations.
Results: Fifty-nine patients (31 boys) were identified. At baseline the median age was 8.2 years (range 4.8–15.8), the mean (SD) for height, weight and body mass index (BMI) for age z-scores were −0.68 (1.31), −0.19 (1.34) and 0.19 (1.38), respectively. At baseline, the mean (SD) z-score for forced expiratory volume in 1 s (FEV1) was −2.61 (1.82). Over 2 years (n = 59), mean FEV1 and forced vital capacity (FVC) improved by 0.17 (95% CI 0.01 to 0.34, p = 0.039) and 0.21 (95% CI 0.04 to 0.39, p = 0.016) z-scores per annum, respectively. Over 4 years there was improvement in height-for-age z-scores (slope 0.05, 95% CI 0.01 to 0.095, p = 0.01) but no improvement in other anthropometric variables. There was no change in spirometry (FEV1 slope 0.00, 95% CI −0.09 to 0.09, p = 0.999 and FVC slope 0.09, 95% CI −0.09 to 0.1, p = 0.859).
Conclusions: Children with non-CF bronchiectasis show adequate growth over time. Lung function stabilises but does not normalise with treatment, underscoring the need for early detection and institution of appropriate therapy.
Statistics from Altmetric.com
Bronchiectasis is conventionally used as a descriptive term for an irreversible pathological state characterised by chronic suppurative airway disease manifested clinically by chronic productive cough and radiologically by bronchial dilation and, often, thick-walled bronchi. The incidence of non-cystic fibrosis (non-CF) bronchiectasis has fallen since the late 19th century, particularly in developed countries,1 and the adult mortality rate associated with it has decreased from over 30% in 1940 to 13% in 1981.2 In children, the disease is reported to have an estimated prevalence of 1 in 5800 in north-east England and 1 in 1700 in New Zealand.1 3 In recent years, diagnosis has become easier with the widespread use of high-resolution computerised tomography (HRCT). Eastham et al recently described a 10-fold increase in the rate of HRCT-diagnosed non-CF bronchiectasis referred to a UK paediatric tertiary hospital.1 The widespread prevalence and increasing diagnosis of this condition in childhood will have a potential impact on adult health services.
In another study, 80% of adults with newly diagnosed bronchiectasis reported chronic respiratory symptoms in childhood, suggesting that the disease process often starts early.2 Unlike adults, in whom bronchiectasis is most commonly reported as idiopathic,4 aetiological factors are identified in more than 70% of some paediatric populations.5 Identifying the underlying aetiology is important, as it allows individualised treatment of a potentially irreversible disease process where early interventions are believed to minimise long-term morbidity and mortality. Recent evidence suggesting that radiological changes described in bronchiectasis may remain static, show improvement or even completely resolve over prolonged periods of time could be due to the beneficial effects of early aggressive treatment.1 6 However, the impact of modern treatment regimens on the course of childhood bronchiectasis has not been well established on long-term follow-up in the developed world.
In many chronic suppurative lung diseases in childhood there is poor growth and a progressive decline in lung function. Although many studies of children with CF have documented the decline in lung function and nutritional issues, far less is known about the course of non-CF bronchiectasis in the developed world. We hypothesised that children with non-CF bronchiectasis could be stabilised with standard aggressive medical treatment. The aim of the study was therefore to evaluate in children with non-CF bronchiectasis: (1) the clinical course of the disease, reflected by change in lung function measurements over a 2- and 4-year period and (2) their growth in the same periods.
METHODS
Subjects
The study population consisted of patients attending the paediatric respiratory clinics at the Royal Brompton Hospital (RBH) and Great Ormond Street Hospital for Children (GOSH) (London, UK) between 1986 and 2002.5 Inclusion criteria were a definitive diagnosis of non-CF bronchiectasis made by HRCT reported independently by paediatric radiologists, the ability to perform reliable spirometry and availability of annual anthropometric and spirometry (forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC)) data for a follow-up period of at least two calendar years. CF was excluded by sweat testing and CF genotype, and by measurement of transepithelial potential difference in equivocal cases. Full details of the diagnostic investigation have been reported elsewhere.5
Patients were reviewed by paediatric respiratory specialists every 3 months in both centres. The treatment protocols did not change during the study and children were prescribed standard aggressive treatment of non-CF bronchiectasis, including airway clearance techniques and oral, nebulised or intravenous antibiotics depending on the nature and frequency of the bacterial isolates. Prophylactic antibiotics were not routinely prescribed and depended on the clinical context. In the absence of any generally agreed protocols and with no evidence base, the decision to start prophylactic antibiotics was made on an individual basis after discussion with the family. The decision was arbitrary and based on a clinical assessment of the severity, frequency and duration of exacerbations. Bronchodilators and steroids were prescribed if clinically indicated, and continued if a response was observed. Specific treatment was also given for any underlying cause (intravenous immunoglobulins for underlying immunodeficiency).5
Study design
A retrospective review of the computerised database previously described was used to identify children eligible for inclusion. Aetiology of bronchiectasis, age, gender, spirometric parameters (FEV1 and FVC), height and weight for each year were recorded. Body mass index (BMI) was calculated as: BMI (kg/m2) = weight in kg/(height in m)2. Z-scores were calculated for all anthropometric measures.7 We analysed children who had 2 years of follow-up data to maximise the sample size, and 4 years to maximise duration of follow-up, albeit at the cost of reducing patient numbers.
Pulmonary function
Spirometry was performed according to the American Thoracic Society guidelines8 with either a Jaeger spirometer at GOSH (Viasys Healthcare, Germany) or a Compact Vitalograph at RBH (Vitalograph, UK) by fully trained paediatric respiratory technicians. At least three technically acceptable manoeuvres were performed each time spirometry was performed, and the highest value of FEV1 and its corresponding FVC were recorded. The flows were reported from the “best” manoeuvre, identified as that with the highest sum of FEV1 and FVC. Data collected were the “best annual” (individual’s best spirometric result for each calendar year) FEV1 and its corresponding FVC. Values were expressed as percentage predicted and z-scores for the patient’s age, sex and height.9 The lung function tests were carried out during routine clinic visits when patients were clinically stable, as far as could be determined. Baseline data were collected during the first patient visit following referral from the local hospital. Children who did not have data for at least one spirometry per year during a period of clinical stability for three consecutive years, were excluded.
Statistical methods
Study size was opportunistic as there were insufficient data to inform a power calculation. Mean and standard deviations were calculated for baseline anthropometric and lung function measurements. Independent sample t tests were used (or the non-parametric equivalent Mann-Whitney test) to compare data between subgroups of different aetiology, FEV1 categories and patient inclusion versus exclusion in 4-year analysis. General Estimating Equations (GEE) were used to describe changes over time; these extend the linear regression model while adjusting for the correlated nature of serial lung measurements.
RESULTS
A presumptive underlying aetiology was identified in 61% of the patients, based on identification or exclusion of a cause using specific investigations (table 1). The baseline mean z-scores for anthropometric measurements were within the normal range (between −2 and +2), but the mean (SD) FVC and FEV1 z-scores were reduced (−2.61 (1.82) and −2.66 (1.68), respectively), confirming obstructive airways disease.
Differences in mean FEV1 and FVC z-scores were noted between various causes of disease (table 2). Children with idiopathic aetiology did not show any difference in spirometric parameters compared with those with primary immunodeficiency, but height-for-age was significantly higher in the idiopathic group (p = 0.003, mean difference 1.06, 95% CI 0.37 to 1.76). The numbers were too small to draw conclusions in the other aetiological groups.
Boys had higher baseline spirometry but the observed differences were not statistically significant (girls: mean (SD) FVC z-score −2.2 (2.02), FEV1 z-score −2.28 (1.82); boys: mean (SD) FVC z-score −3.0 (1.55), FEV1 z-score −3.01 (1.45). There were also no differences in anthropometric z-scores between the two sexes at baseline.
Patients with FEV1 z-scores <−2 at first visit (n = 37) were mainly boys (n = 23) and were significantly older (median age 9.3 years (range 5.8–15), p = 0.03). There were more cases with underlying idiopathic aetiology in this group (43%) than those with normal FEV1 (31%) at baseline. They had a significant lower mean BMI z-score compared with children with FEV1 z-scores of ⩾−2 (p = 0.02, table 3).
Follow-up data at 2 and 4 years are presented. Fifty-nine children (31 boys) with non-CF bronchiectasis had 2-year follow-up lung function data and 31 (17 boys) had 4-year follow-up data (table 4).
During the follow-up period, organisms were isolated in cough swabs or sputum in 36 patients. The two commonest organisms isolated were Haemophilus influenzae (n = 20) and Streptococcus pneumoniae (n = 9), followed by Moraxella catarrhalis (n = 6), Staphylococcus aureus (n = 3), Klebsiella pneumoniae (n = 1) and streptococcus group A (n = 1). Two or more organisms were isolated in 12 children. The most common organisms isolated together were H influenzae and S pneumoniae. During the follow-up period, only one girl had grown Pseudomonas aeruginosa. It was not possible to determine whether these organisms were isolated during infective exacerbations and therefore no further analysis was performed.
Lung function and growth
Three consecutive annual measurements
Serial measurements in 59 patients over a 2-year follow-up period from baseline showed an improvement in FEV1 z-score (slope 0.17, 95% CI 0.01 to 0.34, p = 0.039) and FVC z-score (slope 0.21, 95% CI 0.04 to 0.39, p = 0.016) per annum (fig 1). For example, if a patient started with a baseline FEV1 z-score of 0.8, at 2-year follow-up their expected FEV1 z-score would be 1.14. Anthropometric measurements did not change over the 2-year follow-up period (height z-scores: slope 0.03, 95% CI −0.03 to 0.1, p = 0.274; weight z-scores: slope 0.07, 95% CI −0.01 to 0.16, p = 0.098; BMI z-scores: slope 0.05, 95% CI −0.06 to 0.16, p = 0.335, fig 2).

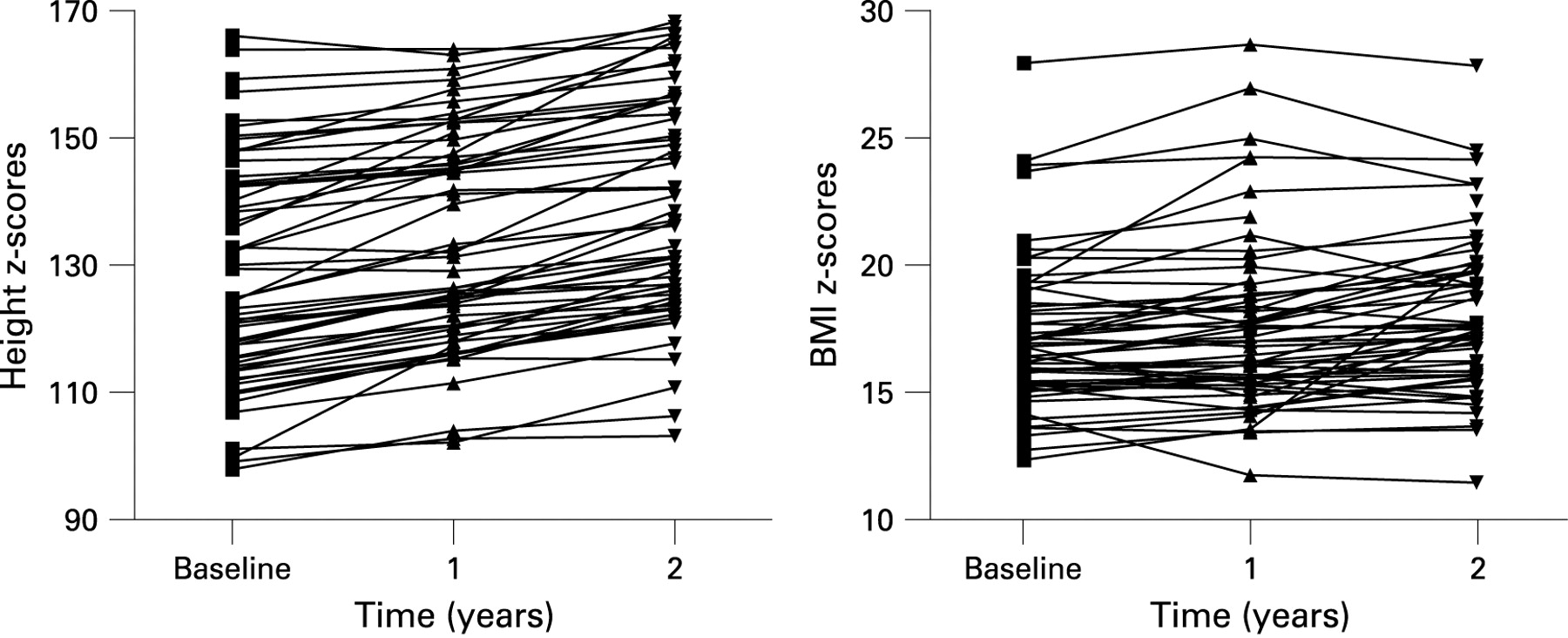
Girls had higher BMI, FEV1 and FVC z-scores with a slope of 0.11, 95% CI −0.58 to 0.89, p = 0.750; 0.34, 95% CI −0.45 to 1.13, p = 0.401; and 0.42, 95% CI −0.41 to 1.24, p = 0.325; respectively, but these were not statistically different. The average height-for-age z-score was lower in girls than in boys by 0.33 z-scores (95% CI −0.98 to 0.31, p = 0.303) over the 2-year period of follow-up.
Those with reduced FEV1 at baseline (<−2 z-scores) also had reduced weight (slope −0.26, 95% CI −0.47 to −0.05, p = 0.017) and BMI z-scores (slope −0.27, 95% CI −0.53 to −0.001, p = 0.049) at 2 years compared with those with normal FEV1 at presentation. Furthermore, the rate of change in spirometry was greater in those with reduced spirometry at baseline (FEV1 slope: −1.84, 95% CI −2.14 to −1.54, p<0.005; FVC slope: −1.8, 95% CI −2.12 to −1.47, p<0.005).
Five consecutive annual measurements
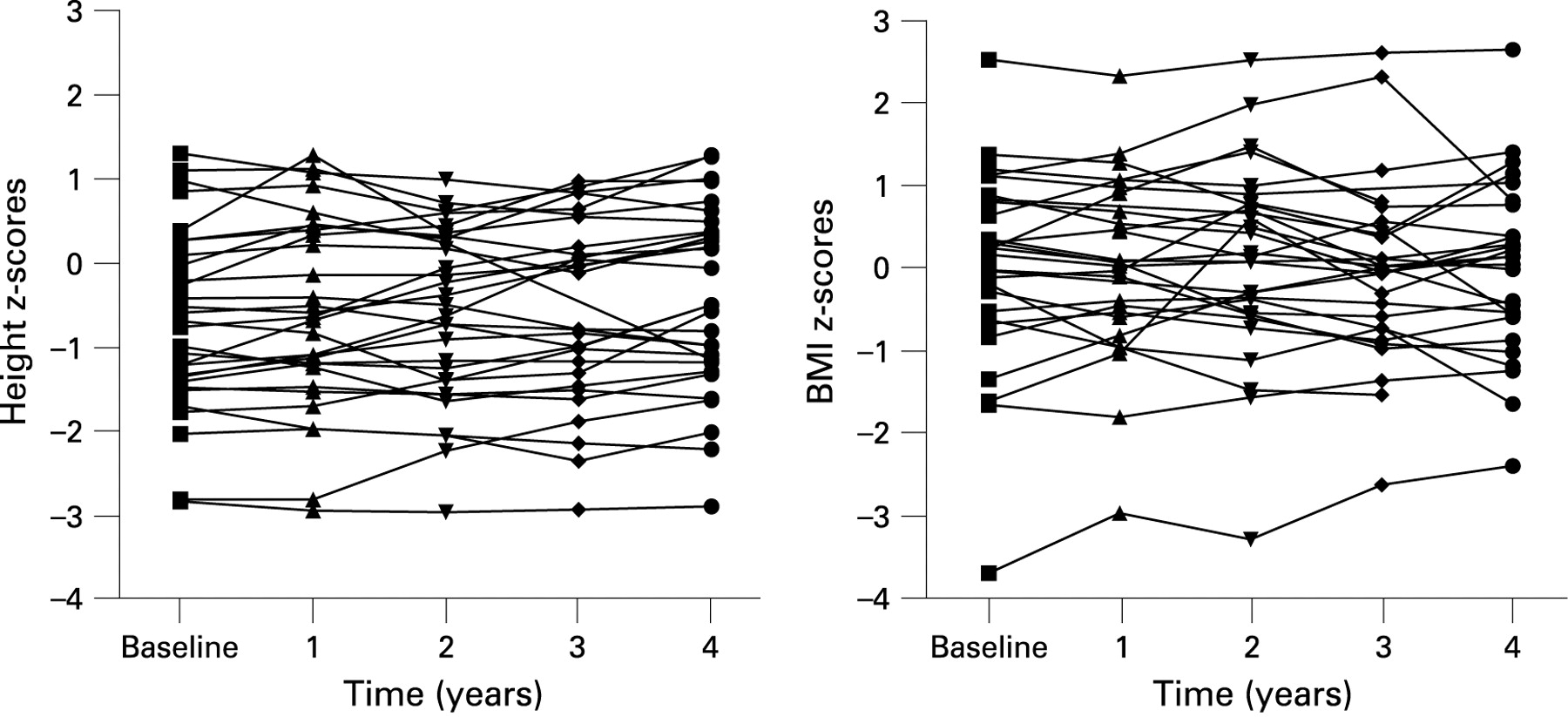
Thirty-one patients were included. The patients excluded were more likely to be older (p = 0.023), have primary immunodeficiency (64%) and less likely to be idiopathic (25%). At baseline there was no difference in mean z-scores for anthropometric or spirometric measurements (table 5).
Examination of five consecutive annual measurements (fig 3) in 31 patients demonstrated an improvement in height-for-age z-score (slope 0.054, 95% CI 0.013 to 0.095, p = 0.01). There were no significant trends for weight-for-age or BMI z-scores. There was also no evidence of change in FVC and FEV1 z-scores (Fig 4) (slope 0.009, 95% CI −0.09 to 0.107, p = 0.859 and slope 0.000, 95% CI −0.09 to 0.09, p = 0.999, respectively. For example, if a patient started with a baseline FEV1 of 0.8 z-scores, at 4-year follow-up their expected FEV1 would 0.84. The group was too small for further subgroup analysis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
The main finding of this study is that children with non-CF bronchiectasis receiving treatment in specialist centres have stable lung function z-scores as determined by spirometry, associated with steady growth (figs 1–4). Children with abnormal lung function at referral were older and had a poorer nutritional state. Although the majority of patients were infected at some time point, we could not characterise their infection status in detail due to lack of data, which is a weakness of this study. Serial examination of the first three consecutive years after initial referral and treatment showed a small improvement in spirometric parameters. In a subgroup in which five consecutive annual data were available, although the mean baseline FEV1 z-score was low, there was no significant deterioration in spirometry or nutrition over the study period. The reasons for the differences in evolution in spirometry over three and five annual measurements are unclear, but inspection of the raw data (figs 1 and 4) suggests that there is no real clinically significant change at any time point, and that spirometry remained stable after referral.
The main limitation of our study is that we have not considered other criteria to assess disease severity such as change in HRCT,10 11 presence of chronic infection, comorbidity or number of exacerbations, which have been shown to be associated with a fall in FEV1 (1.4–3.0% per year) in adults.12 13 The interval between onset of symptoms and diagnosis was not quantified. The follow-up described in our series is limited to the first 2 and 4 years after referral to a tertiary hospital, not after diagnosis. These children were generally referred with an undiagnosed respiratory problem rather than for tertiary management after diagnosis. Other factors known to play a role in growth and lung function evolution are environmental and social issues, which were not studied. It is not routine to repeat HRCT scans on follow-up in children with bronchiectasis at our two centres, hence the lack of HRCT criteria. We cannot exclude the possibility that some children may have shown regression of changes. However, the scans were reviewed by experienced clinicians and radiologists and, as far as is possible, “pre-bronchiectasis” patients were removed from the study. The ideal study, with individual diagnoses studied separately, is not feasible given the low numbers and heterogeneity of paediatric disease. This would require a multicentre study which would introduce more treatment variability. Nevertheless, the study group reflects a cohort of patients with non-CF bronchiectasis in two tertiary respiratory centres. However, it should be noted that these retrospective data with differing aetiologies and overall evolution might not be representative of all patients with non-CF bronchiectasis.
The obstructive lung disease seen in our study is as expected, but conflicting results have been reported regarding its progression in childhood. Twiss et al described deteriorating FEV1 in a group of children aged 6–15 years with non-CF bronchiectasis over a 4.5-year period,14 which was consistent with lung function decline in adults with bronchiectasis.15 16 Ethnicity, social and genetic factors may contribute to these differences as the prevalence of paediatric bronchiectasis in New Zealand is one of the highest reported. In addition, the bronchiectasis described in this population was extensive and severe.10 17 However, Twiss et al also reported a wide variability in individual disease severity and progression, with some children not showing deterioration in their lung function. In contrast, a study from Turkey showed an improvement in FEV1 of 11% per year in school age children.18 Compared with the New Zealand cohort, the children had less severe disease and different management. Our study cannot be directly compared with other similar studies as the clinical context is different, outcome measures are not the same and our spirometry results are presented as z-scores. The use of z-scores is an advantage as they take into account age-related between-subject variability9 so that results over time are more comparable. The extent to which lung function changes over time within the same individual is difficult to assess without knowledge of how much change can be expected in health over the same period.
In the present study, the underlying aetiology for bronchiectasis was known in more than half of the patients (61%). Investigation for a cause of bronchiectasis may allow early implementation of disease-specific therapy (intravenous immunoglobulins) which may have contributed to a good outcome.5 Another possible contributor might be the aggressive use of airway clearance and treatments for infection, but this is conjectural. Antibiotic use was not protocol-driven, which was a weakness of the study, but there is no evidence base or generally agreed consensus on which to base a protocol. There is a clear need for evidence-based guidelines on this point, and the lack of such guidelines makes comparisons between the results of different series difficult.
The study by Twiss et al described a consistent trend for those with primary immunodeficiency to have a more rapid decline in lung function.14 This was not the finding in our study, which included a relatively large group with primary immunodeficiency (n = 25). Although subgroup analysis is not reliable owing to small numbers, the underlying aetiology may influence disease severity. Three patients with post-infectious bronchiectasis had poorer lung function than other groups, with marked airflow obstruction already present at baseline (mean (SD) FEV1 z-score −4.88 (1.37) which is similar to other studies.14 Although post-infectious aetiology is rare in the developed world, it is still a significant cause in developing countries.18 However, it would not be prudent to extrapolate our findings to developing countries, given the very different circumstances—both in terms of aetiology of bronchiectasis and delivery of health care—and the very small numbers of post-infective bronchiectasis in our series.
In contrast to some studies in CF,19–21 most of our patients did not have poor anthropometric parameters at baseline making it impossible to assess if improvement in weight improved lung function. Nevertheless, during the 2-year follow-up period, patients with worse FEV1 had significantly lower weight and BMI z-scores and tended to keep lung function lower. These patients were older at referral and hence were likely to have received treatment later, which might have contributed to the severity of the disease. Overall 2-year mean height z-scores for girls was lower than for boys, but this was not statistically significant (slope −0.33, p = 0.303). Our study shows that girls with non-CF bronchiectasis have equally good lung function and growth as boys, which is similar to recent findings in patients with CF.22
In conclusion, children with non-CF bronchiectasis show adequate growth in time and lung function stabilises but does not normalise with treatment, underscoring the need for early detection and institution of appropriate therapy. Lung function in children diagnosed with non-CF bronchiectasis can be stabilised by modern aggressive treatment, but may nonetheless have considerable respiratory morbidity. We cannot prove from this study that early detection of bronchiectasis and initiation of treatment is beneficial, although this seems likely. Further studies are needed to determine the best evidence-based management for these patients.
REFERENCES
Footnotes
Funding: None.
Competing interests: None.
Ethics approval: Approval was obtained from the local ethics committees of the two hospitals to carry out this retrospective review.
Linked Articles
- Airwaves