Article Text

Abstract
Background: Obstructive sleep apnoea syndrome (OSAS) has been associated with cardiovascular disease in epidemiological and observational studies. Continuous positive airway pressure (CPAP) is the treatment of choice for OSAS, but the impact of this intervention on systemic inflammation involved in the atherosclerotic process remains unclear.
Methods: 100 men with moderate–severe OSAS were randomised to therapeutic (n = 51) or subtherapeutic (n = 49) CPAP treatment for 4 weeks to investigate the effects of active treatment on inflammatory markers such as highly sensitive C reactive protein (hsCRP), interleukin (IL)6, interferon γ (IFNγ) and anti-inflammatory adiponectin.
Results: 4 weeks of therapeutic CPAP did not significantly change blood levels of hsCRP compared with the subtherapeutic control group (difference between median changes −0.24 mg/l (95% CI −0.88 to +0.24); p = 0.30). Plasma levels of IL6 and IFNγ did not change significantly following therapeutic compared with subtherapeutic CPAP (difference between median changes +0.52 and −0.07 pg/ml (95% CI −0.72 to +1.94 and −0.81 to +0.44); p = 0.45 and p = 0.82, respectively). Furthermore, 4 weeks of therapeutic CPAP did not significantly change levels of adiponectin in plasma compared with the subtherapeutic control group (difference between median changes +0.05 pg/ml (95% CI −0.36 to +0.47); p = 0.84). If patients with hsCRP values above 8 mg/l at baseline were excluded, differences between the changes in hsCRP, IL6, IFNγ and adiponectin after 4 weeks of CPAP were smaller, and again not statistically different between groups.
Conclusions: 4 weeks of CPAP treatment has no beneficial effect on blood markers of inflammation and adiponectin in patients with moderate–severe obstructive sleep apnoea.
Statistics from Altmetric.com
The obstructive sleep apnoea syndrome (OSAS) is characterised by repetitive apnoeas/hypopnoeas during sleep, associated with oxygen desaturations and sleep disruption. It has been estimated that between 2% and 4% of the adult population in Western countries suffer from clinically significant OSAS, and it is becoming more prevalent as the average population body weight rises.1
Cross sectional and prospective studies have implicated OSA as an important causal factor in the development of cardiovascular disease.2 The mechanisms underlying the association between OSA and cardiovascular disease are not fully understood. Multiple causal factors leading to vessel wall damage and development of atherosclerotic plaques have been proposed, including reflex sympathetic activation and consequent increases in blood pressure, endothelial dysfunction and systemic inflammation.3–5
Inflammation plays a central role in the initiation and progress of atherosclerosis and has been shown to be involved at the onset of adverse clinical vascular events when activated cells within an atherosclerotic plaque secrete proteases that degrade the fibrous cap, leading to rupture of the plaque and thrombus formation.6 Vascular inflammation involves complex interactions among soluble factors and cells, including proinflammatory cytokines and proteins (eg, interleukin (IL)6, interferon γ (IFNγ), highly sensitive C reactive protein (hsCRP)) and anti-inflammatory cytokines and adipokines (eg, IL10, adiponectin).6 7 Increased levels of inflammatory cytokines and proteins have also been proposed as useful predictors of future cardiovascular events.8 9
Several observational studies have investigated the potential relationship between OSA and hsCRP, proinflammatory cytokines and adiponectin.10–13 Some of these studies found a positive correlation between levels of hsCRP, proinflammatory cytokines, adiponectin and the severity of OSA which was independent of the patient’s obesity.10–13 However, only a few studies, which all included a small number of patients, investigated the effects of continuous positive airway pressure (CPAP) on these pro- and anti-inflammatory markers and have reported conflicting results.14–19 Furthermore, there are no data from randomised controlled trials on the effects of CPAP on proinflammatory cytokines.
To address this uncertainty, we have analysed stored blood samples from a large scale randomised controlled trial on CPAP and measures of cardiovascular risk in patients with symptomatic OSA20 in order to investigate the effects of CPAP treatment on hsCRP, proinflammatory cytokines and adiponectin.
METHODS
Patients
Patients with possible OSA were referred to the Oxford Sleep Unit, Oxford Centre for Respiratory Medicine, UK, by general practitioners, ear, nose and throat surgeons, or other hospital consultants. Patients were eligible for the trial if they were males aged between 20 and 75 years who had excessive daytime sleepiness (Epworth Sleepiness Score ⩾10) and proven OSA with more than 10 oxygen desaturations of >4% per hour (ODI >10/h). All eligible patients were offered participation in the study, unless they required urgent CPAP therapy because of respiratory failure, driving or job issues. The study was approved by the Oxford research ethics committee (COREC No 96.127) and written informed consent was obtained from all participants.
Sleep study, CPAP and assessment of sleepiness
OSAS was diagnosed from a 1 night in hospital respiratory polysomnographic sleep study. Patients’ body movements, heart rate and pulse transit time changes were recorded as measures of arousal from sleep. Pulsoximetry, snoring and increases in the respiratory swing in pulse transit time were used as markers of breathing pattern and respiratory effort (Win-Visi monitoring system; Stowood Scientific Instruments, Oxford, UK) as previously described and validated.21
The results of the sleep study were scored automatically, with manual review to ensure accuracy of the data. OSAS was diagnosed from review of all data and severity was quantified as the number of oxygen desaturations >4% per hour of study (ODI).
After enrolment, patients were randomly assigned to either therapeutic or subtherapeutic CPAP, and then underwent baseline measurements followed by a second sleep study, during which respiratory polysomnography was repeated and CPAP was used according to the assigned group. For patients assigned to therapeutic CPAP, the therapeutic pressure was determined from overnight use of the Sullivan Autoset-T auto-adjusting (ResMed, Abingdon, UK) CPAP machine, from which mask pressure was recorded and synchronised with the sleep study signals, as previously described and validated.22 The record was reviewed the next morning, and the optimum pressure to prevent sleep apnoea, usually the 95th percentile of pressure overnight, was confirmed by a sleep technician. Patients assigned to subtherapeutic CPAP used a machine that delivered <1 cm H2O pressure as previously described,23 which is insufficient to hold the pharynx open.
Patients remained blinded as to whether they were receiving therapeutic or subtherapeutic CPAP, as did the investigators. The sleep nurse, who randomly assigned patients to the two groups, maintained the machines and assisted the patients, was not involved in outcome assessments.
Subjective sleepiness was assessed using the Epworth Sleepiness Score which assesses the tendency to fall asleep during eight typical daytime situations.24 Objective sleepiness was measured with one sleep resistance challenge (Osler test), which tests the ability to stay awake in a darkened and sound isolated room, and was carried out at the same time of day on the two occasions the patient was studied.25
Cardiovascular risk score
A cardiovascular risk score (Framingham Index) was used to objectively assess an individual’s 5 year risk of death due to cardiovascular events.26 The risk score is based on 11 factors including: age, sex, systolic blood pressure, serum total cholesterol concentration, height, serum creatinine concentration, cigarette smoking, diabetes, left ventricular hypertrophy, history of stroke and myocardial infarction. The risk score is an integer, with points added for each factor according to its association with risk. The sum score and corresponding risk of a fatal cardiovascular event were derived from individual patient data according to Pocock et al.26
Inflammatory blood markers
Blood was obtained from all patients in the morning between 09:00h and 11:00h at baseline and follow-up. Measurements of hsCRP, IL6, IFNγ and adiponectin were performed from plasma samples which were stored at −80°C.
The Dade Behring BN method (particle enhanced immunonephelometry, measuring range 0.18–1150 mg/l) was used to measure hsCRP as previously described and validated.27 The internal quality control (Dade Behring Apo-Control-Serum CHD) for measurement of hsCRP has a nominal value of 1.77 mg/l and results are expected to be within the range 1.59–1.95 mg/l. The mean (SD) value of four quality control runs was 1.83 (0.06) mg/l.
IL6 and IFNγ were measured by ELISA with commercially available kits (BD OptEIA; BD Biosciences, San Diego, California, USA). The intra- and interassay coefficients of variation were 4.1% and 10.9%, respectively, for IL6, and 4.0% and 9.9 % for IFNγ. The lower limit of detection for IL6 and IFNγ with these ELISA kits was 1 pg/ml. Samples with IL6/ IFNγ levels <1 pg/ml were re-evaluated (always in pairs of baseline/follow-up samples) by commercially available high sensitivity ELISA kits (BMS213HS and BMS228HS; Bender MedSystems GmbH, Vienna, Austria). The intra- and interassay coefficients of variation were 6.9% and 8.0%, respectively, for IL6, and 6.8% and 7.1% for IFNγ. The lower limit of detection for IL6 was 0.02 pg/ml, and 0.06 pg/ml for IFNγ.
Adiponectin was measured by a commercial ELISA kit (Quantikine Human Adiponectin ELISA; R&D Systems, Abingdon, UK). The intra- and interassay coefficients of variation were 4.7% and 6.9%, respectively. The lower limit of detection was 0.246 ng/ml. All samples were measured in duplicate and in the same batch.
Follow-up
After baseline assessments, patients used their therapeutic or subtherapeutic CPAP machine (Sullivan 6; ResMed, Abingdon, UK) for 4 weeks and then re-attended for repeat measurements of hsCRP, IL6, IFNγ, adiponectin, Epworth Sleepiness Score and the Osler test. Hour meters on the CPAP machines were downloaded to calculate mean nightly use. At the end of the trial, CPAP pressure was re-titrated in every patient to establish their therapeutic pressure for subsequent long term use.
Data analysis
Data are expressed as mean (SD) for normally distributed data and medians (interquartile range) for non-normally distributed data. All statistical analyses were performed with Statistica V.6.0 (StatSoft, Tulsa, Oklahoma, USA). Differences between the groups for the outcomes were assessed by independent t tests if data were normally distributed or by the Mann–Whitney U test if data were not normally distributed. Differences in changes between the groups are expressed as median in the therapeutic, minus median in the subtherapeutic, groups with the 95% confidence intervals (CI) of the difference. Data were analysed on an intention to treat basis, with no change assumed when follow-up hsCRP, IL6, IFNγ or adiponectin data were missing. Data were reanalysed as per protocol, including only those subjects with complete follow-up data to assess whether this differed from the intention to treat analysis. For comparison of frequencies, the χ2 test of independence was used. Spearman’s rank test was used for correlation analysis. A p value <0.05 was considered to be statistically significant.
RESULTS
Trial profile and patients characteristics
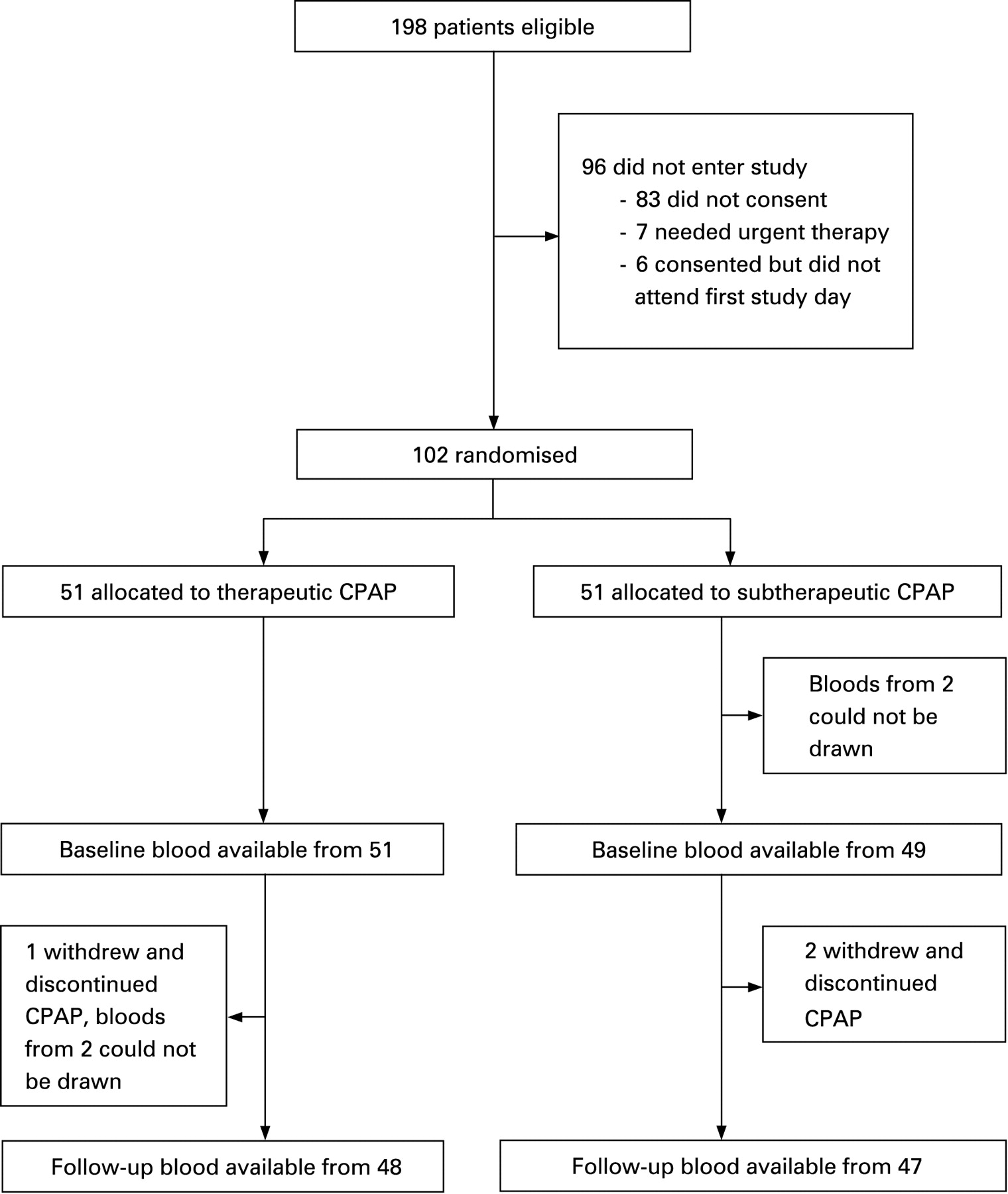
Figure 1 shows the trial profile: 102 patients with a mean age of 48.4 (10.1) years were randomised, 51 to therapeutic and 51 to subtherapeutic CPAP. The two groups were similar in age, body mass index, fat distribution, smoking status, frequency of hypertension (and anti-hypertensive medication), cardiovascular risk score and severity of sleep apnoea (table 1). Blood samples from 100 patients were available for analysis at baseline, and from 95 at follow-up. There were no significant correlations between any of the inflammatory markers and the ODI at baseline.

Data on blood pressure from 52 of the 102 randomised patients had been used in a previously published study evaluating the effect of CPAP on ambulatory blood pressure.23
Measures of sleepiness
Therapeutic CPAP significantly reduced the Epworth Sleepiness Score compared with subtherapeutic CPAP (difference between mean changes −5.3 (95% CI −7.3 to −3.4); p<0.0001), and improved objective sleepiness measured by the Osler test (difference between mean changes +7.7 min (95% CI 2.2 to 13.2 min); p = 0.007). Compliance with CPAP did not differ between the two groups (table 1).
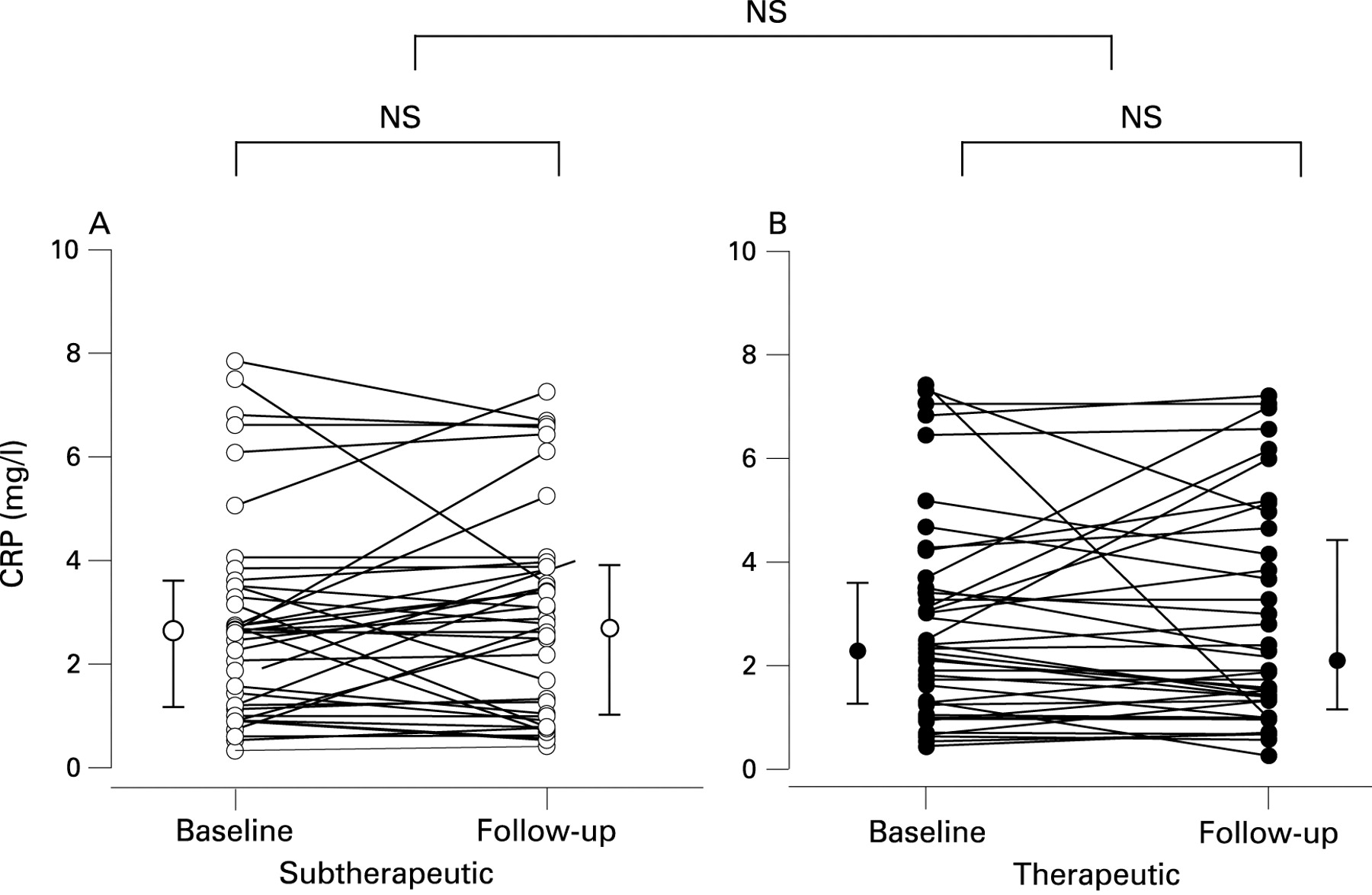
Highly sensitive C reactive protein
hsCRP did not change significantly after 4 weeks of therapeutic CPAP or subtherapeutic CPAP (table 2). If all patients with hsCRP values above 8 mg/l were excluded (10 subjects in each group), which is the threshold for active infection in our institution, the change in hsCRP with therapeutic compared with subtherapeutic CPAP was also insignificant (table 2). Further details on the results are given in table 2, and individual changes in hsCRP are shown in fig 2.
Cytokines
Interleukin 6
Four weeks of therapeutic CPAP did not change plasma levels of IL6 significantly compared with subtherapeutic CPAP (table 2). If patients with hsCRP values above 8 mg/l were excluded, IL6 levels did not significantly change with therapeutic compared with subtherapeutic CPAP (table 2). As would be expected, there was a correlation between IL6 and hsCRP at baseline (r = 0.32 (95% CI 0.13 to 0.49); n = 100, p = 0.001) and at follow-up (r = 0.46 (95% CI 0.28 to 0.61); p<0.0001).
Interferon γ
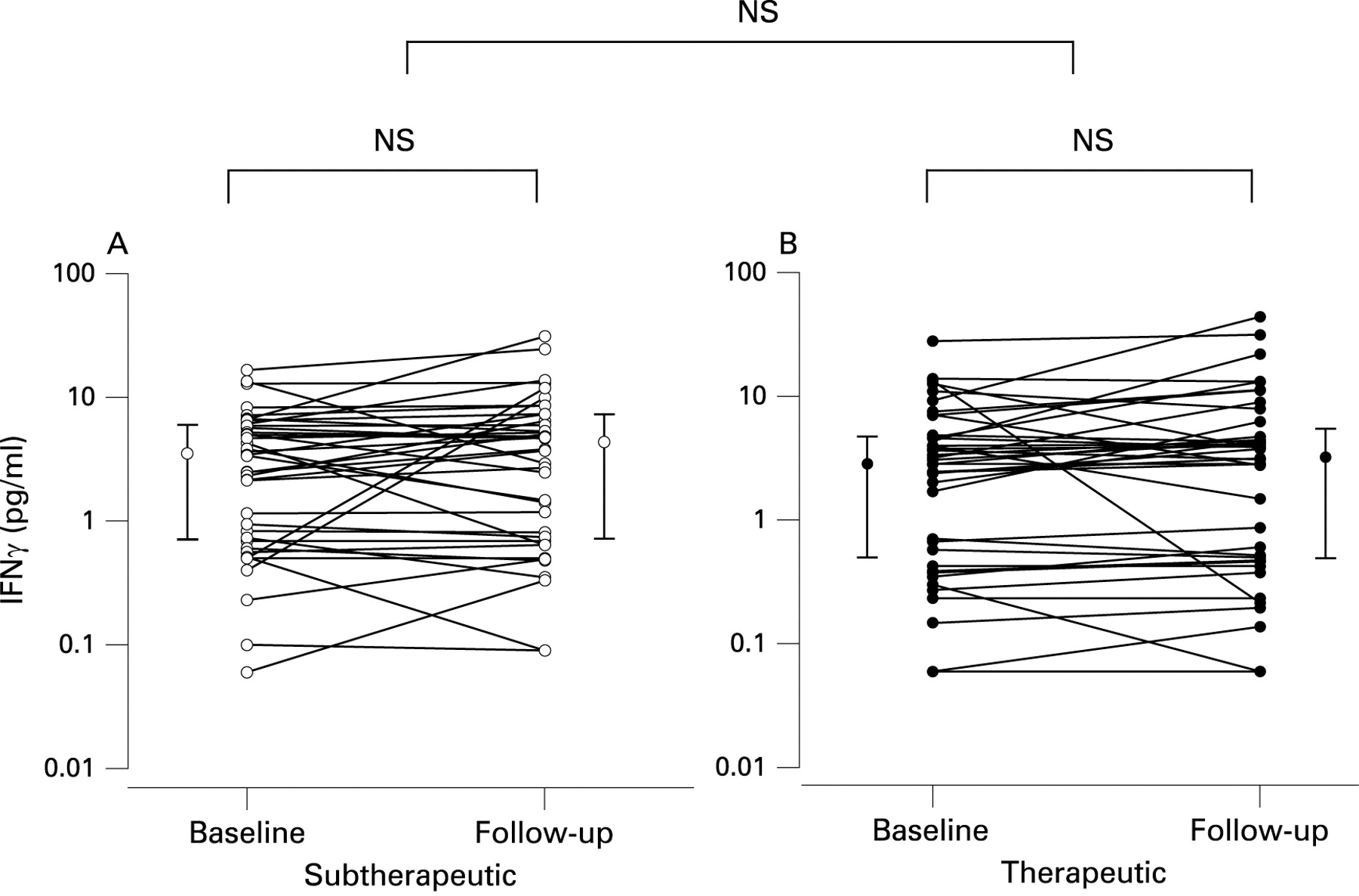
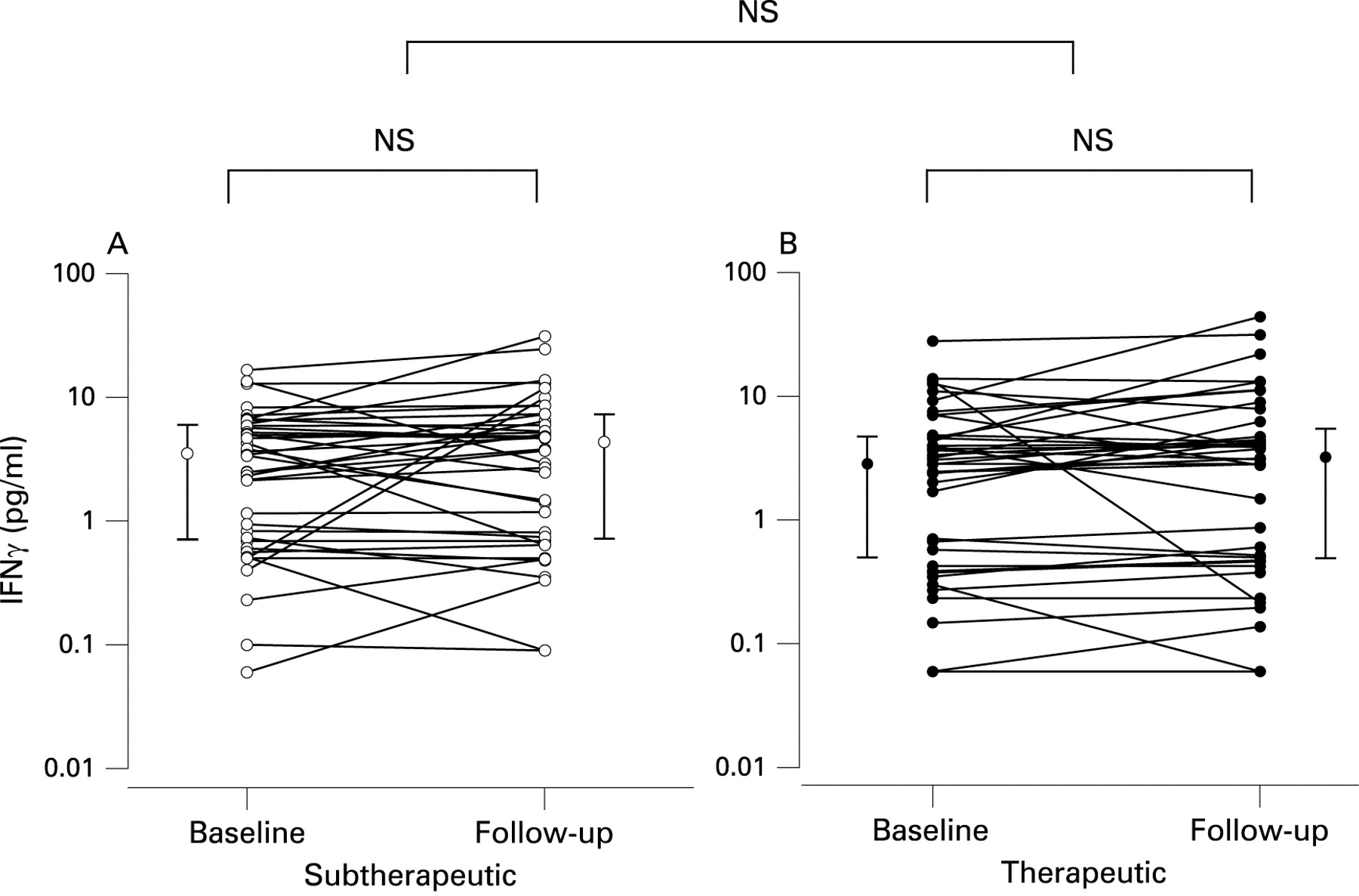
Plasma levels of IFNγ did not change significantly after 4 weeks of therapeutic compared with subtherapeutic CPAP (table 2). If patients with hsCRP values above 8 mg/l were excluded, changes in IFNγ levels were still not significant (table 2).
Further details on cytokine results are given in table 2, and individual changes in IL6 and IFNγ are shown in figs 3 and 4, respectively.

Adiponectin
Four weeks of therapeutic CPAP did not significantly change plasma levels of adiponectin compared with subtherapeutic CPAP (table 2). Individual changes in adiponectin are shown in fig 5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
When data on inflammatory markers were analysed as per protocol, there was no statistically significant difference in changes between the therapeutic and subtherapeutic CPAP groups.
DISCUSSION
We performed a large randomised controlled trial on the effects of CPAP on blood markers of inflammation, hsCRP, IL6, IFNγ and adiponectin, in patients with moderate to severe OSAS, and found no improvement in these parameters after 4 weeks of active CPAP treatment, despite large improvements in both symptoms and objective sleepiness in the group receiving therapeutic CPAP. The 95% confidence intervals of the difference in CRP changes between groups were −0.88 to +0.24 mg/l. Therefore, we have excluded a significant reduction in CRP due to CPAP of more than about 1 mg/l. A power calculation using our data shows that in order to exclude a significant reduction in CRP as small as 0.5 mg/l, with 90% confidence at the 5% level, would require 240 subjects in each arm.
The view that chronic inflammation initiated by monocyte and lymphocyte adhesion to activated endothelial cells plays a central role in the pathogenesis of atherosclerosis is now widely accepted.6 There is also evidence from large epidemiological studies that levels of inflammatory blood markers, such as hsCRP and IL6, are independent predictors of future cardiovascular events and mortality in subjects with and without known cardiovascular disease.9 28 29 Adiponectin, an adipocyte derived peptide, has been shown to have important effects on the cardiovascular system, as reduced plasma levels have been related to insulin resistance, endothelial dysfunction and coronary heart disease.30 31
In the present study, we found that 4 weeks of CPAP did not lower plasma levels of hsCRP in patients with moderate to severe OSA. This finding confirms the negative results we found in a recently published smaller study on the effects of CPAP on hsCRP in patients with newly diagnosed type 2 diabetes and OSA.17 To our knowledge, the effects of CPAP on hsCRP have only been investigated in one other randomised controlled trial. Drager and colleagues32 randomly assigned 24 patients with severe OSA to either CPAP or no treatment, and found a significant decrease in hsCRP after 4 months of CPAP therapy. A possible explanation for the conflicting findings could be that all patients included in the study by Drager and colleagues32 were free of any comorbidities, whereas we included typical OSA patients, some of which had arterial hypertension, diabetes and an increased cardiovascular risk score (table 1). In fact, we found that the chance of a fatal cardiovascular event in the next 5 years was 1.7% in our population compared with the risk of 0.4% for healthy men of similar age26 (ie, a fourfold increase).
The present study is the first randomised controlled trial which has investigated the effects of CPAP on proinflammatory cytokines such as IL6 and IFNγ. We found that 4 weeks of CPAP therapy had no significant effect on plasma levels of these cytokines. Furthermore, levels of proinflammatory IL6 and hsCRP were correlated at baseline and at follow-up (r = 0.32 and r = 0.46, respectively, p<0.001), which strengthens our negative findings. The results of our trial are in concordance with the uncontrolled study of Phillips and colleagues16 who found no change in IL6 levels after short term withdrawal (7 nights) from CPAP. However, Yokoe and colleagues19 found significantly decreased levels of both CRP and IL6 after 4 weeks of CPAP in an uncontrolled study, studying only 17 patients with moderate–severe OSA.
There has been growing interest in the adipocyte derived anti-inflammatory protein adiponectin and its relationship with OSA. There are conflicting reports from observational studies regarding the association of OSA with blood levels of adiponectin, and the results from small uncontrolled studies on the effects of CPAP on adiponectin are not conclusive.33 34 We recently reported that 3 months of CPAP therapy did not increase plasma levels of adiponectin or reduce insulin resistance in patients with type 2 diabetes and OSA.17 In the present study, we found that 4 weeks of therapeutic CPAP did not significantly change plasma levels of adiponectin compared with subtherapeutic CPAP. The reasons for the lack of a change in adiponectin are not clear but may be due to an overwhelming impact of obesity on adiponectin secretion as body mass index was relatively high (35.2 kg/m2) in our study population.
It could be argued that the lack of effect of CPAP therapy on markers of systemic inflammation was caused by the relatively short duration of treatment (4 weeks), although studies on statin therapy have shown a significant reduction of circulatory proinflammatory cytokines in under 7 days following the initiation of therapy.35 However, we cannot exclude the possibility that CPAP treatment over several months might gradually reduce systemic inflammation, and this should be addressed in future longer term randomised controlled trials.
Baseline measurements in our study were performed immediately after randomisation rather than before but, since both the investigators and patients remained blinded to the allocated group, this is unlikely to have introduced a bias.
In conclusion, this double blind randomised controlled trial has shown no improvement in inflammation measured by pro- and anti-inflammatory blood markers in patients with moderate to severe OSA following CPAP treatment, despite symptomatic improvement, as evidenced by large reductions in both subjective and objective sleepiness. The reasons for this are not clear but may be due to the overpowering effects of other cardiovascular risk factors on systemic inflammation. Therefore, we have found no evidence that CPAP produces a cardiovascular benefit through reduction of systemic inflammation in a typical cohort of patients with moderate–severe OSA.
REFERENCES
Footnotes
Funding: ResMed UK made an unrestricted donation to support research work in the Oxford Sleep Unit in 1998 and 2006.
Competing interests: None.
Ethics approval: The study was approved by the Oxford research ethics committee (COREC No 96.127).