Article Text

Abstract
Background: Chronic obstructive pulmonary disease (COPD) is characterised by an abnormal inflammatory reaction of the lungs involving activation of epithelial cells. Leptin is a pleiotropic cytokine important in the regulation of immune responses via its functional receptor Ob-Rb. This study was undertaken to test the hypothesis that severe COPD is associated with increased leptin expression in epithelial cells.
Methods: Immunohistochemistry for leptin was performed on peripheral lung specimens from 20 patients with COPD (GOLD stage 4), 14 asymptomatic ex-smokers and 13 never smokers. Leptin and Ob-Rb mRNA expression were determined by rtPCR in cultured primary bronchial epithelial cells and primary type II pneumocytes. NCI-H292 and A549 cell lines were used to study functional activation of leptin signalling.
Results: Leptin immunoreactivity in lung tissue was observed in bronchial epithelial cells, type II pneumocytes, macrophages (tissue/alveolar) and interstitial lymphocytic infiltrates. rtPCR analysis confirmed pulmonary leptin and Ob-Rb mRNA expression in primary bronchial epithelial cells and pneumocytes. Leptin-expressing bronchial epithelial cells and alveolar macrophages were markedly higher in patients with severe COPD and ex-smokers than in never smokers (p<0.02). Exposure of cultured primary bronchial epithelial cells to smoke resulted in increased expression of both leptin and Ob-Rb (p<0.05). Leptin induced phosphorylation of STAT3 in both NCI-H292 and A549 cells.
Conclusions: Leptin expression is increased in bronchial epithelial cells and alveolar macrophages of ex-smokers with or without severe COPD compared with never smokers. A functional leptin signalling pathway is present in lung epithelial cells.
Statistics from Altmetric.com
Chronic obstructive pulmonary disease (COPD) is a leading and increasing cause of morbidity and mortality worldwide. The airflow limitation in COPD is associated with both structural changes and an abnormal pulmonary inflammatory response to noxious particles or gases, including tobacco smoke.1 Different kinds of inflammatory cells (macrophages, neutrophils, CD8+ T lymphocytes) have been implicated in this chronic inflammation, and a distinct inflammatory pattern has been described in each lung compartment.2 To date, the mechanisms of both recruitment and survival of inflammatory cells in response to noxious particle exposure, which eventually results in a persistent pulmonary inflammation without need of the primary stimulus, remain incompletely understood.
A novel candidate to regulate pulmonary immune function is leptin, a 16 kDa non-glycosylated type I cytokine encoded by the obese (ob) gene.3 Leptin was originally described as an adipocyte-derived hormone involved in balancing food intake and energy expenditure by activating the full-length functional isoform of the leptin receptor (Ob-Rb) in the hypothalamus.4 Recent studies reported leptin secretion not only by adipocytes, but also by various epithelial cell types, as shown for intestinal and gastric epithelial cells,5 6 mammary epithelial cells7 and bronchial epithelial cells.8 The almost universal distribution of leptin receptors reflects the multiplicity of biological effects in extraneural tissues. Recently published studies have shown that leptin has a potentiating role in the function of both innate and adaptive immunity.9 On neutrophils and macrophages, leptin stimulates chemotaxis10 and enhances functional capacities such as oxidative burst,10 phagocytosis11 and cytokine secretion.12 In addition, leptin exerts proliferative13 and anti-apoptotic effects14 on T lymphocytes and promotes Th1 cell differentiation.15 Furthermore, leptin enhances the host response to inflammation and infection by stimulating tissue repair via its mitogenic and angiogenic properties on epithelium and endothelium.16 17
Normal lung tissue displays particularly high levels of Ob-Rb,18 19 and specific leptin binding sites have been identified in airway and alveolar epithelial cells.20 21 This designates the lung as a peripheral site of action for leptin in pulmonary diseases. It has recently been shown that leptin protein is actually present in induced sputum22 and proximal airway biopsies8 of patients with COPD. We therefore hypothesise that COPD is associated with increased leptin expression in lung epithelial cells. To this end, we investigated leptin protein in peripheral lung tissue from patients with severe COPD, ex-smokers with normal lung function and never smokers. In addition, we analysed the effect of smoke on leptin and Ob-Rb mRNA expression using primary bronchial epithelial cell (PBEC) cultures. NCI-H292 and A549 cell lines were used to study functional activation of leptin signalling.
METHODS
Full details are given in the supplementary material available online.
Selection of subjects
Human lung tissue was obtained from 20 patients with stable COPD (GOLD stage 4) admitted to the pulmonary rehabilitation centre CIRO Horn, Horn, The Netherlands for 6 weeks before undergoing lung volume reduction surgery (LVRS) and 27 control subjects (6 current smokers, 8 ex-smokers, 13 never-smokers) with normal lung function who underwent a lobectomy or pneumectomy for lung cancer at the University Hospital Maastricht and University Hospital Ghent to obtain tissue blocks for immunohistochemistry;23 9 anonymous patients who underwent a lobectomy or pneumectomy for lung cancer at the Leiden University Medical Center, Leiden, The Netherlands to isolate PBEC for in vitro experiments; and 3 lung transplant recipients to isolate primary alveolar type II pneumocytes for in vitro experiments (see online supplement). The study was approved by the medical ethical committees of the University Hospital in Maastricht, Ghent and Leuven. All subjects gave written informed consent.
Immunohistochemistry
Paraffin sections (4 μm) were processed for immunohistochemical analysis of leptin, CD68 and thyroid transcription factor-1 (TTF-1). A validated protocol for sequential double staining was used to identify leptin-positive macrophages. Sections were incubated with anti-leptin (Santa Cruz Biotechnology, Santa Cruz, California, USA) and visualised using Blue substrate kit III (Vector Laboratories, Burlingame, California, USA). Consecutively, sections were incubated with anti-CD68 (Dako Cytomation, Glostrup, Denmark) and developed using Red substrate kit I (Vector Laboratories). A similar protocol was used to identify leptin-positive alveolar type II pneumocytes using anti-TTF-1 (Lab Vision, Fremont, USA) and DAB substrate (Dako Cytomation).
Cytoplasmic immunostaining for leptin was examined in PBEC, tissue macrophages and alveolar macrophages. At least 500 cells were counted by one independent observer unaware of the clinical data and expressed per cell type as the percentage leptin-positive cells of the total counted cells.
Cell culture
Human PBEC
Subcultures of PBEC were cultured either submerged (n = 4 subjects) or at an air-liquid interface (ALI; n = 5 subjects). Cigarette smoke condensate (CSC) was prepared using commercial cigarettes (Caballero, British American Tobacco Group). Cultures were stimulated with 2 AU/ml CSC for 24 h (submerged) or 1-5-10 AU/ml CSC for 48 h (ALI; apically) and medium was stored at −20°C. RNA was isolated from submerged and ALI cultures and stored at −80°C until further use.
A549 and NCI-H292 cell lines
A549 and NCI-H292 cells were serum-deprived for 18–24 h followed by stimulation for 0.5–1–2 h in 0.5% FBS-containing medium: (a) without additions; (b) 20–100–500 ng/ml recombinant human leptin (R&D Systems); (c) 50 ng/ml recombinant human IL-6 (PeproTech, Rocky Hill, NJ, USA). Cell lysates were stored at −80°C until rtPCR or Western blotting analyses.
Quantitative real-time PCR
Total RNA was isolated using the RNeasy Microkit/Minikit (QIAGEN, California, USA) and reverse transcribed into cDNA (Reverse-iT 1st strand Synthesis Kit; Abgene, Epsom, UK). Primers for leptin, Ob-Rb, PPIA, HPRT-1 and HSP-90B are listed in the online supplement. PCR reactions were performed on an iCycler iQ Real-Time PCR system (BioRad, Hercules, California, USA) using SYBRgreen dye (BioRad). The standard curve method was used to calculate the relative quantity of the respective genes. The geNorm applet (http://medgen.ugent.be/∼jvdesomp/genorm/) was used to calculate a normalisation factor based on the expression of three reference genes (PPIA, HPRT-1, HSP-90B).
Leptin ELISA
Leptin levels in basal medium of PBEC cultured at ALI were determined using a commercially available human leptin immunoassay (Quantikine DLP00; R&D Systems, Minneapolis, USA) with a lower detection limit of 7.8 pg/ml.
Western blotting
Whole cell lysates were separated by SDS-PAGE (reducing conditions) and transferred to nitrocellulose. Blots were incubated with antibodies raised against phospho-STAT-3 or total-STAT-3 (Cell Signaling, Beverly, Massachusetts, USA). Immunoreactive proteins were visualised using chemoluminescence (Pierce Biotechnology, Rockford, Illinois, USA).
Statistical analysis
Results are presented as mean (SD) for normally distributed variables and median (range) otherwise. Non-parametric data were compared by the Kruskal-Wallis test followed by the Mann-Whitney U test as appropriate. The χ2 test was used to compare categorical variables. Correlations between parameters were evaluated using Pearson rank correlation analysis (SPSS Version 13.0, SPSS, Chicago, Illinois, USA). A p value of <0.05 denotes the presence of a significant statistical difference.
RESULTS
Subject characteristics
The clinical and lung function characteristics of the study subjects are summarised in table 1. Patients with COPD had decreased forced expiratory volume in 1 s (FEV1; % predicted), ratio of FEV1 to forced vital capacity (FVC) and carbon monoxide lung transfer factor (Tlco; % predicted) and increased residual volume (RV; % predicted). The COPD and ex-smoking control groups were similar with regard to age, gender and body mass index (BMI). The ratio of current to ex-smokers was significantly different in the COPD group than in the asymptomatic smoking group, whereas the smoking history (pack-years) was not. The never smoker group was matched for age and BMI but consisted of considerably more women.
Leptin immunostaining in peripheral lung
Leptin immunostaining was observed in lung tissue of every subject examined. In patients with severe COPD, leptin staining was most pronounced in lung epithelium, especially in the cytoplasm of the BECs at the apical (luminal) side (fig 1A). Consistent staining was noted in the cytoplasm of alveolar type II pneumocytes (TTF-1-positive) in peribronchiolar and perivascular areas, and in alveolar walls with interstitial fibrosis. Leptin staining was also noted in (infiltrating) inflammatory cells. Intense staining was detected in anthracosis pigment-containing tissue macrophages (fig 1D) and a subset of CD68+ alveolar macrophages (fig 1C). Lymphocytes situated in interstitial lymphocytic infiltrates (fig 1E) showed weaker and more diffuse staining for leptin. Pulmonary vessels showed no specific staining (fig 1F). Comparable staining pattern and intensity was observed in current smokers, ex-smokers and never smokers with normal lung function, with one exception: alveolar macrophages from never smokers mostly showed membranous staining for leptin and a less intense cytoplasmic staining (fig 1G).

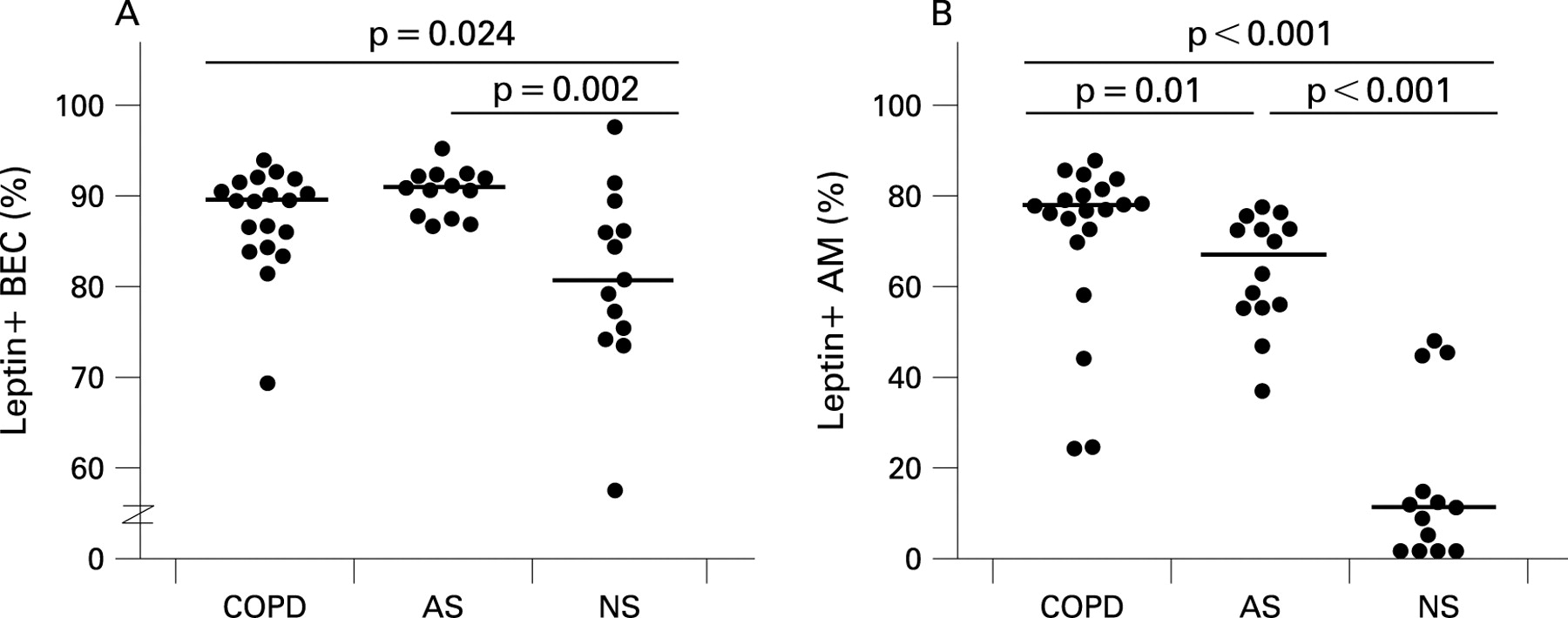
Subsequent quantitative analysis of leptin-positive cells was performed in BEC, alveolar macrophages and tissue macrophages on sections sequentially double-stained for leptin and CD68, allowing distinction between alveolar macrophages and alveolar type II pneumocytes in alveolar walls. Owing to the small amount of cytoplasm of TTF-1-positive alveolar type II pneumocytes (especially in control subjects) combined with the sometimes weak staining in these subjects, TTF-1-positive/leptin-positive double-stained alveolar type II pneumocytes could not be quantified. As shown in fig 2, the counts for leptin-positive BEC were significantly higher in patients with COPD (median 89.3%) and asymptomatic smokers (median 90.7%) than in never-smokers (median 80.7%; p<0.05). In addition, patients with severe COPD had significantly more leptin-expressing alveolar macrophages (median 77.2%) than asymptomatic smokers (median 66.3%; p = 0.01). The counts for leptin-positive alveolar macrophages were even lower in never smokers (median 11.4%; p<0.001). Tissue macrophages containing anthracosis pigment showed 100% positivity for leptin in all subjects examined (including never-smokers). No significant differences were noted between ex-smokers and current smokers or between men and women.
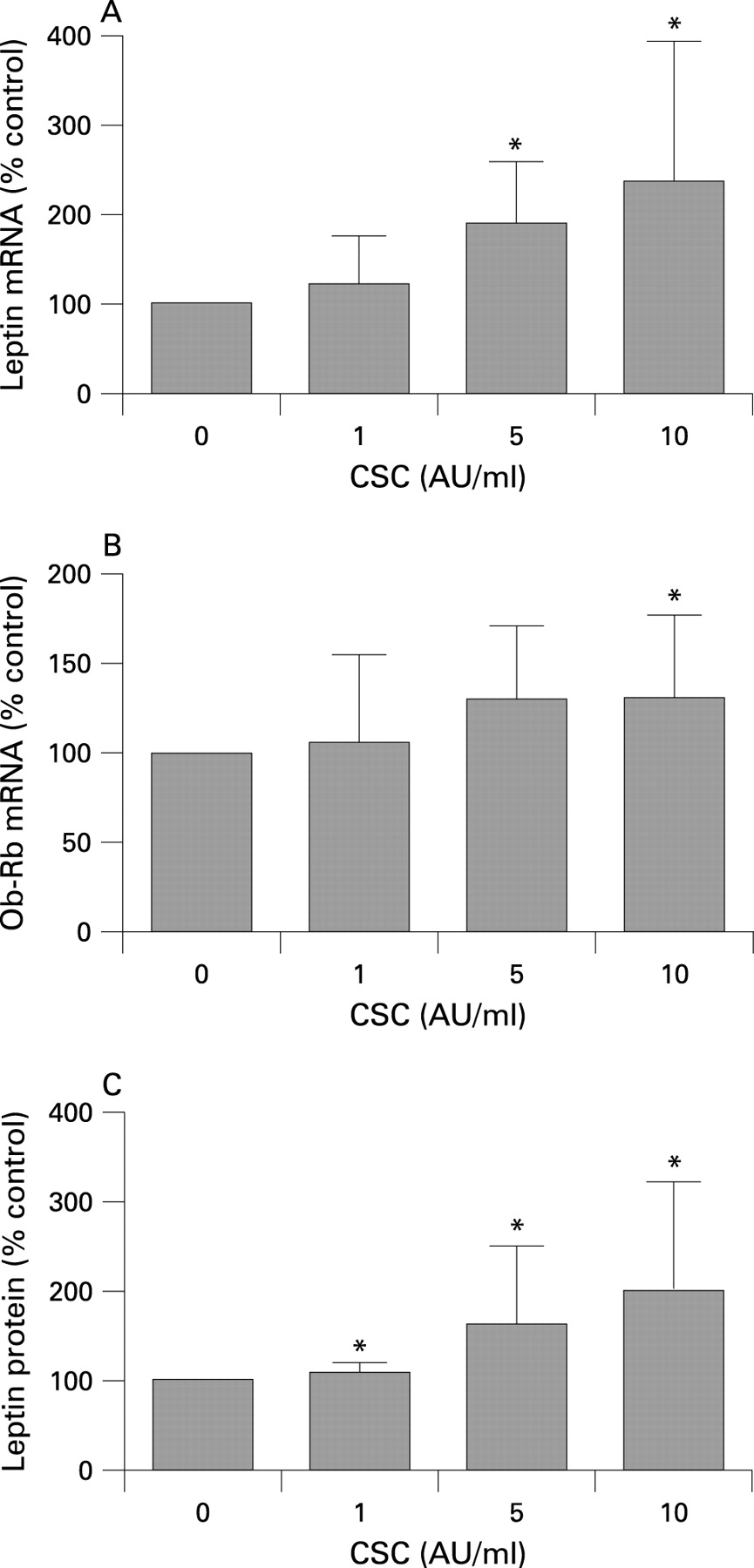
Leptin and Ob-Rb mRNA expression in cultured primary epithelial cells and cell lines
To confirm expression of leptin and its functional receptor Ob-Rb in human lung epithelial cells, rtPCR analysis was performed on cultured PBEC, primary alveolar type II pneumocytes and the NCI-H292 and A549 cell lines. First, PBEC cultures were isolated from nine donors and cultured submerged (n = 4) or at an ALI (n = 5). Basal leptin and Ob-Rb mRNA expression was detected in all PBEC cultures. As the immuno-data strongly suggest that smoking itself may induce leptin expression, PBEC cultures were stimulated with increasing doses of CSC. Stimulation of submerged PBEC cultures with 2 AU/ml CSC resulted in increased leptin mRNA expression in three out of four donors (mean increase 128% of untreated cultures). A dose-response experiment performed in the ALI cultures showed a significant induction of leptin mRNA expression after 24 h stimulation with 5–10 AU/ml CSC (fig 3A); Ob-Rb mRNA expression showed a similar pattern (fig 3B). Measurements of leptin protein in basal medium by ELISA strengthened the mRNA data, demonstrating constitutive leptin protein production (14.4 pg/ml) and a significant dose-dependent increase after stimulation with CSC (fig 3C). Leptin mRNA expression and protein production in ALI cultures showed a strong correlation in the individual samples examined (R = 0.941, p<0.001). Second, primary alveolar type II pneumocytes purified from lungs of lung transplant recipients showed mRNA expression for leptin and Ob-Rb (see online data supplement). Third, both NCI-H292 and A549 cells expressed leptin and Ob-Rb mRNA (data not shown), which is in line with the results of the primary cultures (fig 3 and fig E1 in online data supplement). Constitutive mRNA expression levels of both leptin and Ob-Rb were approximately fivefold higher in A549 cells than in NCI-H292 cells. These data suggest that both BEC and alveolar type II pneumocytes are a source of leptin in the human lung and can potentially respond to leptin due to expression of the functional leptin receptor isoform Ob-Rb.
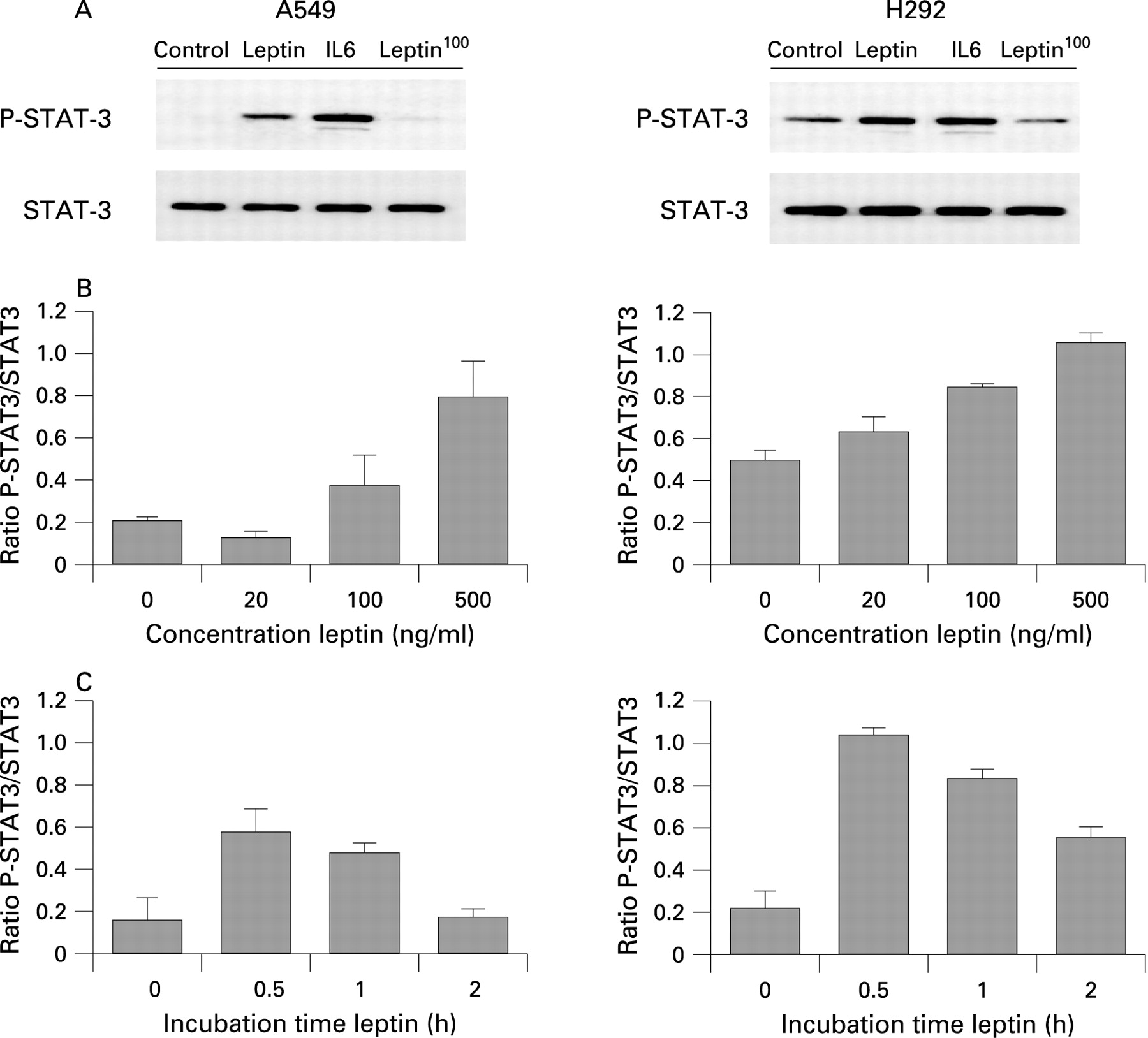
Leptin-induced STAT-3 phosphorylation in lung epithelial cell lines
The functionality of Ob-Rb in NCI-H292 and A549 cells was evaluated by stimulating cells with leptin and determining the phosphorylation status on tyrosine 705 of STAT-3 by Western blot analysis. Figure 4A shows that STAT-3 is constitutively phosphorylated in NCI-H292 cells but not in A549 cells. Leptin stimulation induced phosphorylation of STAT-3 in both cell lines, with interleukin-6 stimulation as a positive control and heat-inactivated leptin as a negative control. Leptin stimulation induced phosphorylation of STAT-3 in a dose-dependent (fig 4B) and time-dependent (fig 4C) manner in both cell lines. Together, these results show that bronchial epithelial cells and alveolar type II cells can respond to leptin, resulting in activation of JAK/STAT, the classical signal transduction pathway of leptin.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
Leptin is increasingly recognised as a pleiotropic cytokine important in regulating food intake and energy expenditure, reproduction, haematopoiesis, angiogenesis and immune responses. In this light, we previously reported increased circulating leptin levels in patients with emphysema related to the systemic proinflammatory status and dietary intake.24 We also showed that leptin protein levels in induced sputum are highly correlated with inflammatory markers (tumour necrosis factor α (TNFα) and C-reactive protein) in patients with mild to moderate COPD.22 More recently, Bruno and colleagues8 reported leptin expression in proximal bronchial biopsy specimens of smokers with or without COPD. This study extends the latter results to the peripheral compartment and investigated expression and localisation of leptin on protein and mRNA levels in peripheral lung tissue from ex-smoking subjects with and without severe COPD and never smokers. In addition, smoke-induced expression of leptin was investigated using ALI cultures of PBEC. Functionality of leptin signalling was investigated using in vitro cell culture models.
This study provides the first evidence that BEC, alveolar type II pneumocytes and macrophages are a source of leptin in human peripheral lung. Immunohistochemical analysis showed a strong cytoplasmic staining for leptin in BEC at the apical site and quantification showed significant differences between never-smokers and smokers, whereas differences between asymptomatic smokers and patients with severe COPD were less obvious. These data strongly suggest that smoking itself induces leptin expression in BEC. Using ALI cultures of PBEC, we were able to confirm this hypothesis, showing that increasing doses of CSC induced mRNA expression of leptin and its functional receptor Ob-Rb, and secretion of leptin protein to the basal medium. Our observation that distal bronchiolar epithelium is capable of producing leptin is in line with data from Bruno et al8 who noted positive immunostaining for leptin protein in the central airway epithelium of bronchial biopsy specimens. In contrast, Bruno et al8 showed decreased leptin-positive epithelial cells in smokers and patients with COPD compared with never smokers, which may be explained by the fact that different tissue specimens were used in the two studies. As recently demonstrated for Toll-like receptor 4 and β-2-defensin, the central airway epithelium may behave differently from the distal airway epithelium in patients with COPD.25 Epithelial leptin expression may be further affected by the inflammatory pattern, which is known to be clearly different in central versus peripheral airways.26 Further studies are needed to study differences in epithelial cell behaviour throughout the bronchial tree.
In addition to BEC, we noted clear staining for leptin protein in TTF-1-positive alveolar type II pneumocytes. In addition, leptin mRNA expression was demonstrated in freshly isolated and cultured alveolar type II pneumocytes from patients with severe COPD (see online supplement). Taken together, these data strongly suggest that both BEC and alveolar type II pneumocytes are a source of leptin in the human lung.
Next to lung epithelium, leptin protein was demonstrated in alveolar macrophages and in interstitial tissue macrophages. Quantification of leptin-positive alveolar macrophages showed an increase in ex-smokers compared with never smokers, and a further increase in patients with severe COPD. The difference in the ratio of current to ex-smokers in the study groups makes it difficult to evaluate whether the differences between groups can be attributed to the presence/absence of COPD or to the presence/absence of a subgroup of current smokers. Leptin expression in macrophages has previously been described in lymph nodes and acute/active inflammatory lesions in the brain and spinal cord in murine experimental autoimmune encephalomyelitis.27 Whether macrophages are indeed capable of expressing leptin or whether the immunoreactivity of these macrophages is due to leptin uptake remains to be studied. Our preliminary data indicate, however, that leptin is detectable in alveolar macrophages in bronchioalveolar lavage fluid (unpublished results).
Leptin exerts its functions through Ob-Rb, and this study confirms abundant Ob-Rb expression in human total lung, PBEC, primary alveolar type II pneumocytes, NCI-H292 cells and A549 cells. Moreover, functionality of Ob-Rb signalling in lung epithelial cells was demonstrated by a time- and dose-dependent increase in phosphorylation of the transcription factor STAT-3 in both A549 and NCI-H292 cells after stimulation with leptin. Our observation that pulmonary leptin expression differs between patients with severe COPD, asymptomatic smokers and never smokers—while BMI does not—lends further support for the local production of leptin in the lung independent of metabolic parameters. The question as to which role leptin fulfils in the lung is therefore of great interest. Many studies indicate potent immunomodulating effects of leptin, which may also be true for pulmonary leptin. Systemic leptin was shown to protect against TNFα-induced toxicity,28 suggesting that leptin is involved in protective mechanisms that allow an organism to cope with potentially autoaggressive effects of its immune system. In line with these findings, Mancuso et al showed that leptin-deficient mice exhibit impaired host defence in bacterial pneumonia.11 29 30 In contrast, leptin is also known as a neutrophil survival factor,8 and intratracheal instillation of leptin caused lung oedema and injury.21 Besides its possible role in immunomodulation, leptin may also act as a growth factor in the lung,19 contributing to epithelial repair and regeneration in COPD.31 In any case, the presence of Ob-Rb in the epithelium and immune cells32 indicates that pulmonary leptin is a potentially important mediator, allowing cross-talk between resident lung epithelial cells and immune cells in response to noxious particles.
One limitation of our study is the control groups used. The study groups were carefully matched for gender, age, BMI and smoking status (pack-years smoked), which have been recognised as potential confounders.33–35 In contrast to patients with severe COPD from whom lung tissue was obtained during LVRS, all control subjects had primary lung cancer adjacent to the macroscopic normal lung tissue studied, raising the concern that leptin expression may have been directly affected by the adjacent tumour. Although only those specimens without any histological abnormalities (apart from different stages of emphysema and alveolar macrophages with smoker’s pigment) were selected, we cannot exclude the possibility that our data are biased by the presence of the tumour. We addressed this limitation by checking gene expression for leptin and Ob-Rb in (cultured) primary lung epithelial cells in which the influence of a nearby tumour can be excluded, and by performing additional in vitro stimulation experiments using well characterised bronchial and alveolar epithelial-like cell lines.
In summary, our study provides the first evidence that BEC, alveolar type II pneumocytes and macrophages are a source of leptin in human peripheral lung tissue. We demonstrated increased numbers of leptin-positive BEC and leptin-positive alveolar macrophages in smokers with and without severe COPD compared with never smokers. In addition, leptin expression in primary BEC could be induced by stimulation with smoke, indicating that leptin may be important in smoking-induced pathology. Furthermore, bronchial and alveolar epithelial cells were shown to express the functional leptin receptor isoform Ob-Rb and to respond to leptin, resulting in activation of the JAK/STAT signalling pathway. Detailed studies are currently in progress to determine functional target genes of leptin signalling in lung epithelial cells and to elucidate the action of leptin in the lung, which may help in understanding the role of lung epithelial cells in the pathogenesis of COPD.
Acknowledgments
The authors thank Dr H J Pennings (CIRO Horn, Horn, The Netherlands) and Dr I K Demedts (Department of Respiratory Diseases, Ghent University Hospital, Ghent, Belgium) for their help in recruitment of the patients; Dr P H Hoet (Laboratory of Pneumology, Unit Lung Toxicology, K U Leuven, Leuven, Belgium) for performing the in vitro experiments involving primary alveolar type II pneumocytes; Dr S van Wetering (Department of Pulmonology, Leiden University Medical Center, Leiden, The Netherlands) for performing the in vitro experiments involving submerged PBECs; Greetje Castelijns (Department of Respiratory Medicine, University Hospital Maastricht, Maastricht, The Netherlands) and Coby van Run (Department of Pathology, University Hospital Maastricht, Maastricht, The Netherlands) for technical assistance with the immunohistochemical stainings.
REFERENCES
Supplementary materials
web only appendices 64/1/26
Footnotes
▸ Additional details are published online only at http://thorax.bmj.com/content/vol64/issue1
Funding: This study was supported by research grants of the Netherlands Asthma Foundation (3.2.05.046), Concerted Research Action of the University of Ghent (BOF/GOA 01251504) and AstraZeneca, The Netherlands.
Competing interests: None.
Ethics approval: The study was approved by the medical ethical committees of the University Hospital in Maastricht, Ghent and Leuven. All subjects gave written informed consent.