Article Text

Statistics from Altmetric.com
Some patients with cystic fibrosis (CF) may be symptom-free when well but many will have chest symptoms such as cough and sputum production. Antibiotic treatment is prescribed on the basis of new symptoms or worsening of existing symptoms, hence the term “exacerbation” is used. Pulmonary exacerbations in patients with CF have an important negative effect on quality of life1 and survival,2 and prompt diagnosis with effective multidisciplinary management is therefore essential. There have been attempts to establish clear criteria for the diagnosis of pulmonary exacerbations, notably in a consensus document produced by the US CF Foundation.3 There is, however, a lack of agreement among clinicians about important diagnostic features, with differences between those factors used by paediatricians and physicians.4 This is not surprising; for example, preschool children with CF may wheeze rather than cough, may not produce sputum and will not be able to perform pulmonary function tests. It is important to diagnose and treat pulmonary exacerbations promptly in this age group. A prospective validation of the US CF Foundation criteria has identified the most reliable indicators for a pulmonary exacerbation (compared with the “gold standard” of a physician diagnosis).5 These are: decreased exercise tolerance; increased cough; increased sputum; absence from school or work; increased adventitial sounds on lung examination; and decreased appetite. Adding change in lung function to this list has little effect on sensitivity and specificity. Antibiotic treatment of pulmonary exacerbations is guided by identification of specific organisms from the airways.
INFECTION IN EARLY LIFE
From the first weeks of life, patients with CF may have intermittent or chronic infection of the lower respiratory tract with a number of organisms. Bronchoalveolar lavage in infants at a mean age of around 3 months has shown that approximately one-third had infection with Staphylococcus aureus in the lower respiratory tract.6 A bronchoscopy study on a larger group of older children at a mean age of 17 months found S aureus in 47%, Haemophilus influenzae in 15% and Pseudomonas aeruginosa in 13%.7 The presence of lower respiratory infection in young children with CF is associated with more frequent symptoms (such as wheeze), increased levels of inflammatory mediators and increased air trapping.8 In contrast, children in whom infection is successfully treated show a fall in the level of inflammatory mediators to pretreatment levels.9 However, in CF there is inflammatory dysregulation, with inflammatory markers (such as interleukin-8 and neutrophils) showing a more vigorous response than in non-CF individuals.10
EXACERBATIONS AND PAERUGINOSA
Most patients with CF in the UK have chronic pulmonary infection with P aeruginosa by their late teens.11 Infection with P aeruginosa is associated with a more rapid decline in lung function and an increase in mortality.12 P aeruginosa has the ability to establish chronic infection through biofilm formation on damaged respiratory epithelium, and this allows chronic infection to occur as well as contributing to antibiotic resistance.13 However, the symptoms of an exacerbation may be due largely to the release of more sensitive planktonic organisms from the biofilm layer.14 In young children there is often considerable genetic heterogeneity in isolates of P aeruginosa.15 This contrasts with adults chronically infected with P aeruginosa in whom pulmonary exacerbations are usually not caused by a new strain of this organism. In a study which identified the strain of P aeruginosa by genotype, the same genotype was found during an exacerbation and during a period of stability in 94% of patients.16
Many patients with CF will develop a pulmonary exacerbation in the presence of chronic lower respiratory infection, often with P aeruginosa. In many cases there will be an additional factor which has triggered an exacerbation in a vulnerable patient, and identifying this factor or factors may be the key to effective management of the exacerbation. Table 1 gives some possible precipitating factors, their diagnostic features and a summary of their management.
Parents and patients may fail to give important maintenance treatment because they are overwhelmed or because they have not fully understood its importance. Rarely, even with appropriate social support and interventions from the multidisciplinary team, essential treatment is still not given and, in these circumstances, child protection procedures should be followed. Where non-adherence to maintenance treatment such as inhaled antibiotics and airway clearance has led to an exacerbation in an adult, it is essential to have an open dialogue. Once the pulmonary exacerbation has been treated, it may be helpful for the physician and patient to agree which maintenance treatment is essential and to attempt to adhere to this reduced programme. As exacerbations may still occur even with optimum adherence, it is important to reassure parents and patients to continue maintenance therapies.
Respiratory syncytial virus (RSV) is the most common cause of bronchiolitis in young infants and infants with CF are vulnerable to more severe infection. The monoclonal antibody palivizumab may be administered monthly during the winter season to prevent RSV infection, but there is no clear recommendation in favour of this at present.17 Treatment is supportive with a low threshold for intravenous antibiotics, as RSV infection has been linked with the acquisition of infection with P aeruginosa.18
Influenza A can cause a severe pulmonary exacerbation in patients with CF.19 Influenza immunisation is frequently recommended, although good evidence of benefit is lacking.20 Antibiotic and supportive treatment is essential and the use of antiviral agents such as zanamivir has been reported.21
The most common fungal complication in CF is allergic bronchopulmonary aspergillosis (ABPA), the features of which may be similar to those of a pulmonary exacerbation. It has been proposed that, for patients with CF, ABPA is likely if there is a fourfold rise in the level of IgE accompanied by positive aspergillus precipitins in the presence of clinical and radiological deterioration.22 The prevalence of ABPA in UK CF clinics is reported to be around 6%.23 Treatment is with oral prednisolone 0.5–1 mg/kg once daily for 2–3 weeks followed by the same dose alternate daily for 2–3 months if there is improvement in clinical and radiological features.24 There is some evidence that adding itraconazole will reduce the frequency of recurrence of ABPA and may have a steroid sparing effect (table 2).25
Mucus plugging may lead to segmental or lobar collapse (fig 1). This must be treated vigorously as a collapsed lobe will become severely bronchiectatic. Initial treatment with intravenous antibiotics and physiotherapy targeted at the collapsed lobe should be augmented with dornase alfa (if the patient is not already receiving this) and prednisolone to reduce mucosal oedema. If this is unsuccessful, flexible bronchoscopy with suction under direct vision should be undertaken. When bronchoscopy is undertaken under general anaesthesia through an endotracheal tube, care must be taken to avoid the mucus plug completely blocking the tube (fig 2). Dornase alfa may also be instilled under direct vision.28 In some cases blind bronchial lavage may be undertaken by a physiotherapist, instilling 0.9% saline, once a patient has been anaesthetised for bronchoscopy.


Infection with a group of organisms collectively termed “atypical mycobacteria” may cause the symptoms of a pulmonary exacerbation in CF. The most common organisms in the atypical group are Mycobacterium avium complex (72%) and Mycobacterium abscessus (16%). Overall the prevalence of this group of organisms is thought to be around 13%.29 Patients with this infection tend to be older and to have relatively good lung function. Infection is less common in patients with P aeruginosa. Guidelines for diagnosis have been suggested by the American Thoracic Society.30 Treatment is prolonged (12 months), using three drugs, started sequentially, with combinations determined by the specific organism (table 1).31
TREATMENT OF EXACERBATIONS
Many patients with CF will experience an exacerbation with no new organism or obvious precipitant. Most of these will have chronic pulmonary infection with Gram negative organisms such as P aeruginosa, Burkholderia cepacia complex or Stenotrophomonas maltophilia. Many CF clinicians will treat with anti-pseudomonal antibiotics during an exacerbation if the patient has had previous pulmonary infection with P aeruginosa, even if it is assumed that this has been eradicated. Other patients may have had frequent isolates of other common organisms such as S aureus and should be treated according to the most recent sensitivities. However, the management of pulmonary exacerbations in patients with chronic P aeruginosa without any obvious precipitating factor presents a number of dilemmas:
-
Whether to give intravenous antibiotics.
-
Whether to use one intravenous antibiotic or more than one.
-
Which antibiotics to choose.
-
How frequently, and for how long, should antibiotics be administered.
There is only one class of anti-pseudomonal antibiotics which is active orally—the quinolones. Ciprofloxacin (table 2) is the most commonly used drug in this class in CF. A resistance rate to ciprofloxacin of 30% has been reported in strains of P aeruginosa isolated from patients with CF in the UK.32 If the oral route is used, then the only option for administering a second antibiotic is in nebulised form (colistin or tobramycin). The choice of intravenous antibiotics need not require a hospital admission. Similar results can be achieved with home intravenous treatment provided appropriate patients are selected and adequate community support for patients and families is available.33
Numerous studies have demonstrated improvements in symptoms, forced expiratory volume in 1 s, lung function, exercise capacity and markers of inflammation following treatment with intravenous antibiotics. However, a number of other interventions may occur in parallel with antibiotics such as intensification of airway clearance treatments.
A small study has compared an intravenous anti-pseudomonal antibiotic (ceftazidime) with placebo (both groups received physiotherapy and other supportive care) for pulmonary exacerbations of CF in patients over 12 years of age. There was little difference between the two groups in the numbers of patients rated as “improved”, although there were more dropouts in the placebo group.34 The only other study to compare an intravenous antibiotic with placebo for exacerbations (in children with CF) recorded two deaths and fewer patients with improved lung function in the placebo group.35
Many patients with CF who have received multiple courses of antibiotics will develop problems with intravenous access, making intravenous antibiotic treatment difficult or impossible. In these circumstances, the early placement of an indwelling intravenous access device allows intravenous treatment to be given in a timely fashion. However, these devices may result in complications (see below).
Combination anti-pseudomonal antibiotics have been recommended because of concern that monotherapy may be associated with increased levels of antibiotic resistance.36 A systematic review of single versus combination antibiotic therapy found that studies were of poor quality, and no significant differences could be found in efficacy or safety. However, there was a non-significant trend towards increased levels of antibiotic resistance at 2–8-week follow-up with single compared with combination antibiotic therapy.37
Where two antibiotics are chosen, they should have a different mechanism of action (where possible). A β-lactam-based antibiotic and aminoglycoside combination is frequently used. There has recently been interest in the possibility of studying the effects of antibiotics when used in combination (synergy testing) and using this as a guide to the choice of antibiotics. This might be of particular value in infections with multiply resistant P aeruginosa. However, this has not been shown to result in an improved clinical response when the results of synergy testing conducted prior to the exacerbation (as opposed to during the exacerbation) are used.38
Where B cepacia complex has been identified in respiratory secretions, antibiotics such as meropenem and ceftazidime have been shown to have some in vitro activity39 while temocillin has been shown (in an uncontrolled study) to have some clinical benefit.40 Cotrimoxazole has also been used and may be given orally.41 For sensitive strains of S aureus, intravenous flucloxacillin (which may be combined with tobramycin) may be used. For methicillin resistant S aureus (MRSA), intravenous teicoplanin is convenient (once daily). For oral treatment, the antibiotic sensitivity pattern may allow doxycycline to be used (adults and older children) or linezolid (a more expensive alternative). While the use of these antibiotics may improve symptoms, MRSA is rarely eradicated. Stenotrophomonas maltophilia is an emerging pathogen in CF and has in vitro sensitivity to doxycycline (adults and older children) and colistin. Intravenous colistin is also effective against most strains of multiresistant P aeruginosa (table 2).32
A large randomised controlled trial conducted in adults and children has shown that the total daily dose of tobramycin may be administered once daily rather than in three divided doses as has been common practice in CF. Efficacy was the same in both regimens and there was less nephrotoxicity with once daily treatment in children.42 Similarly, the pharmacokinetics of ceftazidime suggest that it would be more effective given as a continuous infusion. However, studies of continuous versus intermittent ceftazidime have been underpowered and have failed to show any difference between regimens.43 A minimum duration of antibiotic treatment of 10 days has been recommended.24 Many centres give 2 weeks of treatment as a routine. There is no evidence from randomised studies as to the optimal duration of treatment.
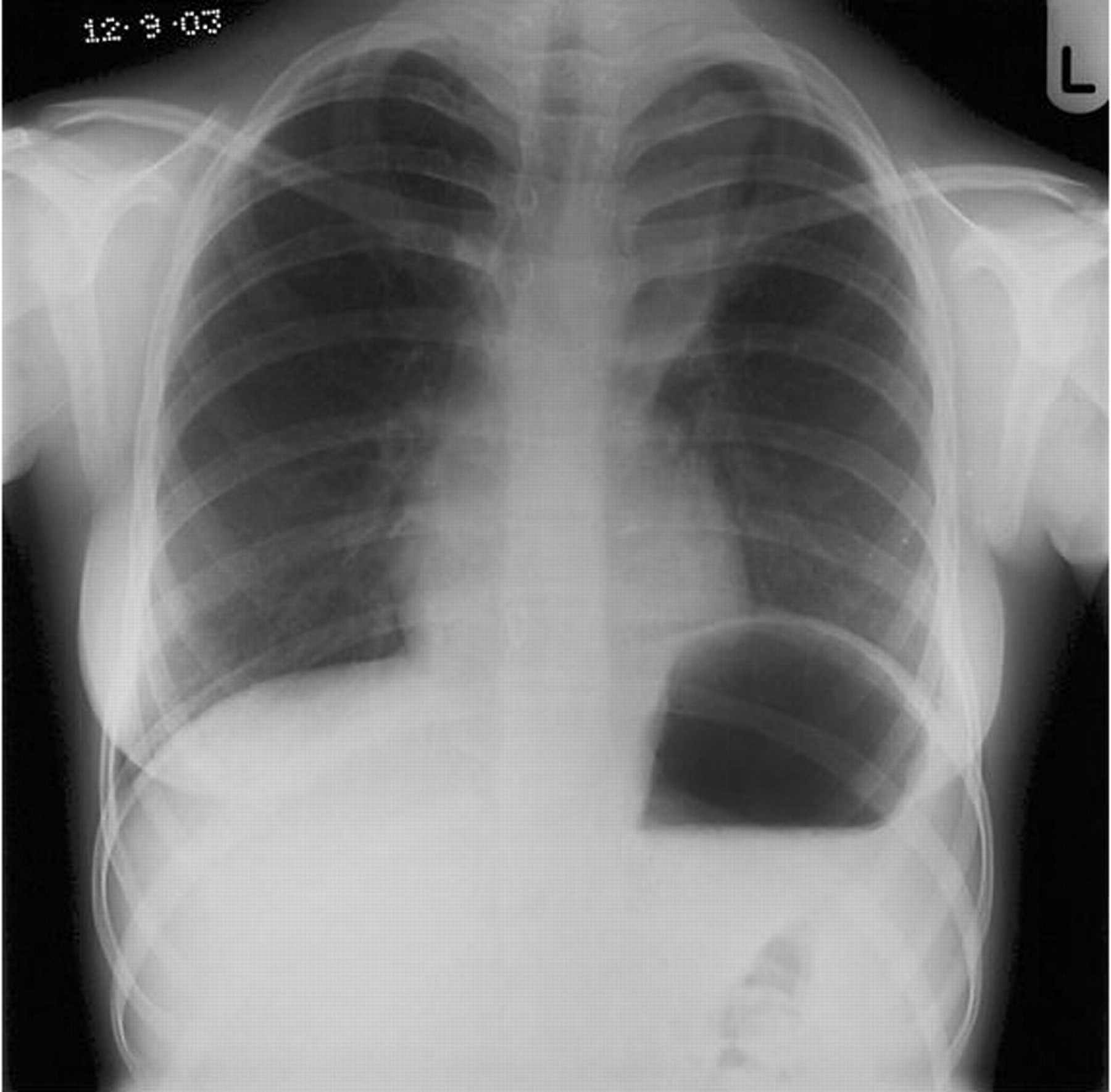
Most patients with CF die of respiratory failure.44 The first episode of respiratory failure may start as a pulmonary exacerbation in individuals with severe disease. Non-invasive ventilation may be helpful in patients with respiratory failure in the short term. Ventilation with endotracheal intubation is associated with poor outcome when used as an emergency, although survival is better in children under 5 years of age.45 Areas of severe bronchiectasis may develop into a pulmonary abscess requiring prolonged antibiotic therapy and even percutaneous drainage (fig 3). In patients with an indwelling intravenous access device who develop high spiking fevers while on intravenous antibiotics, infection of the access device should be considered. Candida is one of the most serious pathogens found in these circumstances and may be treated with intravenous amphotericin (table 2). However, even with appropriate antifungal treatment, removal of the access device is often necessary.

{kind=link}
{kind=link}
{kind=link}
The appropriate use of antibiotics is only part of the package of care needed for patients with CF suffering from a pulmonary exacerbation. A range of chest physiotherapy techniques may be appropriate, including percussion and postural drainage, positive end expiratory pressure techniques and occasionally the use of the Bird ventilator. Patients with CF may become catabolic during an exacerbation and energy requirements will be increased. However, appetite may be diminished and swallowed sputum may lead to vomiting, particularly after gastrostomy or nasogastric feeds. The careful combination of airway clearance and a gradual increase in nutritional support may help to overcome this problem. The short-term use of oral steroids during a pulmonary exacerbation may be beneficial, but good evidence is lacking.
CONCLUSIONS
Early recognition and vigorous management of pulmonary exacerbations in the patient with CF is crucial to the maintenance of lung function, good quality of life and survival. The aetiology of an exacerbation in each patient should be carefully considered. A multidisciplinary approach with attention to airway clearance and nutrition, as well as appropriate antibiotic treatment, is essential.
Acknowledgments
The authors acknowledge the help of Susan Clarke, Cystic Fibrosis Pharmacist at Nottingham City Hospital.
REFERENCES
Footnotes
-
Funding: None.
-
Competing interests: AS has received research funding from Forest Laboratories and has provided consultancy advice to Profile Pharma.