Article Text

Statistics from Altmetric.com
From the question on page 1082
The lung biopsy showed a picture consistent with a congenital surfactant deficiency (either of protein B or C) or the absence of lamellar bodies in alveolar type 2 cells (fig 1).
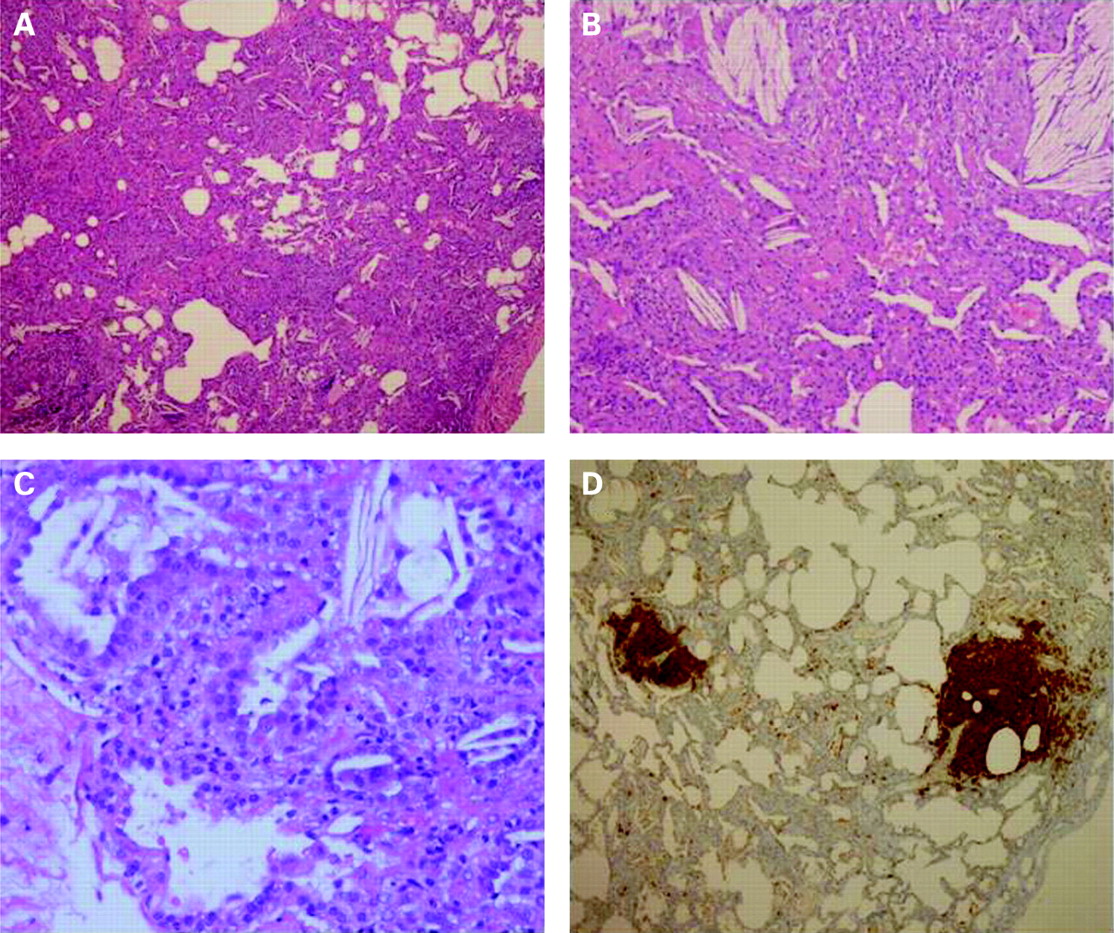
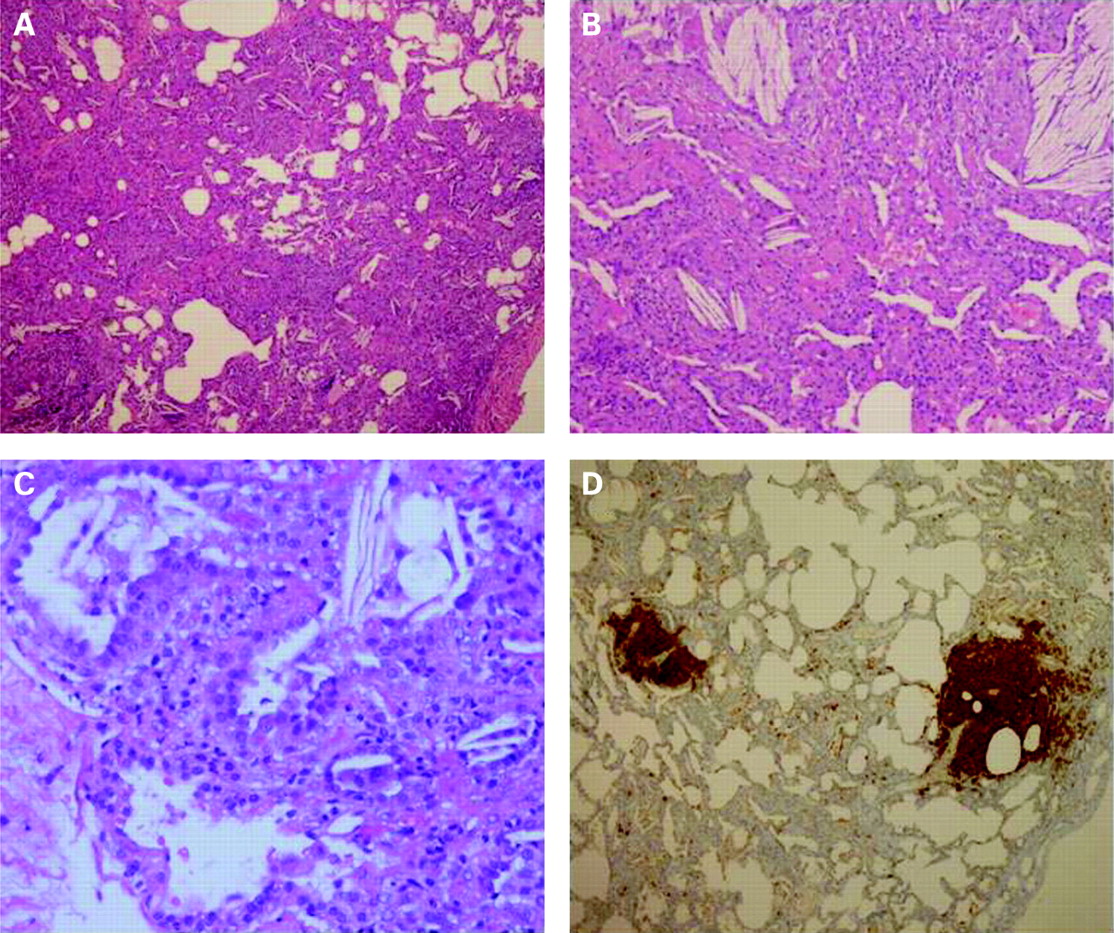{kind=link}
Genetic analysis showed a de novo mutation in SFTP-C gene (I73T aminoacidic substitution). Diagnosis of surfactant protein C deficiency (SP-C) was formulated.
Hydroxychloroquine 10 mg/kg/day was started in the first twin, preceded by 3 months of prednisone 2 mg/kg/day because of the severity of the disease. As they began treatment, only one episode of upper airway infection occurred. After 18 months of continuous hydroxychloroquine treatment the respiratory rate at rest was 36/min and 34/min, respectively, and oxygen saturation 99%. The levels of physical activity were normal for age, digital clubbing was no longer evident and height raised back to 25° centile.
Surfactant protein C deficiency
Mutations in SFTP-C gene cause certain forms of interstitial lung disease which present both as recurrent lung infection and chronic hypoxaemia, cyanosis, clubbing and growth failure.1 Moreover, I73T mutation is associated with an interfamilial phenotypic variability. Surfactant C seems to play an important role as a hydrophobic protein in stabilisation of surfactant. In heterozygous mutations the aberrant protein interacts with the regular pathway of regular SP-C biosynthesis, leading to inhibition of functionally active SP-C production. Lung damage is therefore induced both by SP-C deficiency in the alveolus and by aberrant proSP-C that cannot be handled by the protein degradation pathway and induces cellular injury and pulmonary inflammation.
In these patients hydroxychloroquine treatment seemed effective. Since prednisone was also used in one of the twins, a possible benefit from the combination of the two drugs can also be suggested. A worsening of the clinical condition after interruption of treatment would be needed to firmly confirm efficacy, even though the long lasting clinical improvement was remarkable.
The exact mechanism of action of hydroxychloroquine is unknown. Antimalarial agents have profound effects on macrophages by interfering with antigen presentation, by inhibiting production of inflammatory mediators such as interleukin (IL)1 and IL6 and by inhibiting activation of toll-like receptors. In SP-C deficiency the drug may act not only through an anti-inflammatory action but also by blocking the intracellular processing of SP-C precursors.2 To our knowledge, only very few cases of SP-C deficiency successfully treated with hydroxychloroquine have been reported.3 4
SP-C deficiency should be suspected in any case of unusually severe recurrent lung infections or undiagnosed interstitial lung disease in infants.