Article Text

Abstract
Aim: A multicentre retrospective study was undertaken to examine patients with interstitial lung disease (ILD) with the initial clinical manifestation of an anti-synthetase syndrome (anti-Jo-1 antibodies), and to analyse the characteristics and long-term outcome of these patients according to their clinical presentation (acute or gradual onset), treatment and adverse events related to treatment.
Methods: 32 patients, 15 (47%) presenting with acute onset and associated respiratory insufficiency (group A) and 17 (53%) with gradual onset (group G) were examined. Myositis was diagnosed at admission in only 31% of cases and was observed during follow-up in 56% of cases, but the prevalence did not differ between the two groups.
Results: Fever and radiological patterns including diffuse patchy ground-glass opacities, basal irregular lines and consolidation on high-resolution CT scan were more frequent in group A than in group G. More patients in group G had neutrophils in the bronchoalveolar lavage fluid and autoantibodies other than anti-Jo-1 (rheumatoid factor, anti SSa/SSb) than in group A. The percentage of patients in whom the ILD improved at 3 months was significantly higher in group A than in group G (13/15 vs 9/17; p = 0.006). In contrast, after 12 months, most patients with ILD progression were in group A and were treated with corticosteroids alone. A combination of corticosteroids and an immunosuppressive drug was required in most cases (84%) at the end of the follow-up period. Severe adverse effects of treatment were observed and varicella zoster virus infection was frequent.
Conclusions: Early testing for anti-synthetase antibodies, particularly anti-Jo-1, and creatine kinase determination are useful procedures in patients presenting with ILD. Treatment with corticosteroids and immunosuppressive drugs is required in most patients. At the end of the study, around two-thirds of patients had stable ILD while the other third had disease progression with respiratory insufficiency.
Statistics from Altmetric.com
Interstitial lung disease (ILD) may be associated with systemic inflammatory disorders and autoantibody production. The development of ILD has been described in detail in patients with polymyositis and dermatomyositis,1–10 and has been shown to increase morbidity and mortality in patients with myositis.11–15 Anti-synthetase antibodies, including anti-Jo-1 antibodies (antihistidyl-tRNA synthase), are found in 20–30% of patients with myositis and are most frequent in those with polymyositis.2 7 16–21 However, Schmidt et al, in a large series of patients, showed that the clinical picture of the anti-Jo-1 antibody syndrome is variable; indeed, most patients have features other than myositis at disease onset, including ILD.20 Anti-Jo-1 antibodies have rarely been detected in patients with ILD as the sole manifestation of autoimmune disease.20 22–24
A study was undertaken to evaluate a large number of patients with anti-Jo-1 antibodies who presented with ILD as the initial clinical manifestation, with or without extrarespiratory symptoms suggestive of connective tissue disease, and to describe the characteristics and long-term outcome of these patients according to their clinical presentation (acute or gradual onset), treatment and adverse events related to treatment.
METHODS
Study design and patient selection
This multicentre study was conducted in 12 pulmonology departments of university hospitals from the “Assistance Publique des Hôpitaux de Paris” and Lille University Hospital. These teaching and tertiary care centres were contacted by email and asked to identify patients with ILD and anti-Jo-1 antibodies at initial evaluation from their registers. Only patients with clinical and radiological findings consistent with ILD, anti-Jo-1 antibodies and a follow-up of at least 12 months before the study were included.
Patients who had previously been diagnosed as having polymyositis, dermatomyositis or inclusion body myositis and who had been treated before lung involvement occurred, as well as those with dysphagia and/or a subsequent episode of aspiration pneumonia, were excluded from the study. However, patients with myositis (increase in creatine kinase (CK) and/or aldolase, myalgia) as well as arthalgia, Raynaud’s phenomenon or skin manifestations before the respiratory symptoms occurred were not excluded from the study if these conditions were the initial manifestations of the anti-Jo-1 syndrome.
Data collection
One physician (ITL) used a standard form to record the characteristics of patients from medical files and contacted the pneumology consultant. The data collected on the form included epidemiological characteristics, ILD characteristics, extrapulmonary manifestations on admission and outcome data evaluating the progression of ILD and extrarespiratory manifestations (at 3 months, 1 year and at the end of the follow-up period), survival and adverse events related to treatment.
Characteristics of ILD on admission
Clinical symptoms assessed were cough, dyspnoea (NYHA) and haemoptysis. Crackles on auscultation were reported. Patients were assigned to two groups: acute (A) and gradual (G), based on an analysis of the histogram of distribution of the duration of onset (time in weeks between the first symptom and the diagnosis of anti-Jo-1 syndrome) and the presence or absence of acute respiratory failure. Blood gas analysis in ambient air, carbon monoxide transfer factor (Tlco), total lung volume (TLV), slow vital capacity (SVC) and forced expiratory volume in 1 s (FEV1) were reported.
Particular attention was paid to high-resolution computed tomography (HRCT) scans of the lung which were reviewed and interpreted by two researchers (ITL and JC). The following characteristics were assessed and noted on a standard form:25 irregular linear opacities (irregular thickening of interlobular septa), septal thickening (abnormal visualisation of interlobular septa), consolidation, ground-glass opacities, honeycombing, traction bronchiectasis and bronchiolectasis, pleural effusion and adenopathy. The predominance and location were described (eg, lower lobe predominance, peripheral predominance).
Bronchoalveolar lavage (BAL) fluid was analysed and the results of cell component (differential counts) and microbacteriological analyses were recorded. Normal reference values26 are: <15% lymphocytes, <5% neutrophils and <1% eosinophils. Transbronchial biopsy and surgical lung biopsy results analysed according to the classification of Katzenstein and Myers25 were recorded.
Characteristics of extrapulmonary manifestations on admission
Weight loss, fever (>38°C), asthenia, cutaneous manifestations, arthralgia, arthritis, Raynaud’s phenomenon, myalgia and muscle weakness were recorded. CK levels were recorded and values more than twice the upper limit of the normal range (2N) were considered abnormal. Electromyography (EMG) results were recorded. Myositis was defined as CK >2N and/or an EMG result consistent with the diagnosis of myositis and/or muscle weakness before corticosteroid treatment.12
Outcome data for ILD and extrarespiratory symptoms
The outcome data for ILD and extrarespiratory symptoms were evaluated at 3 months, 12 months and at the last date of follow-up. Treatment regimens (corticosteroids, immunosuppressive drugs) and changes in treatment during follow-up were recorded. The response to treatment of ILD was evaluated according to the ATS criteria.27 Improvement, stability or failure to respond was documented based on symptoms, HRCT scan and lung function test.
All adverse effects related to treatment were recorded. Deaths were also recorded and classified according to whether they were attributable to the disease, treatment or neither.
Statistical analysis
All results were expressed as median (range). Comparisons were made by the non-parametric Mann-Whitney test and the Fisher exact test.
RESULTS
Characteristics of the population at initial evaluation (n = 32)
All participating hospitals responded, but 5 of the 13 centres did not have any eligible cases. A total of 49 cases of ILD with anti-Jo-1 antibodies were reported between 1 January 1994 and 31 May 2004, of which 17 were excluded (10 because myositis was diagnosed and treated before ILD, 2 due to recurrent aspiration pneumonia and 5 because of inadequate follow-up). Thirty-two patients were therefore evaluated by indirect immunofluorescence on Hep2 cells (cytoplasmic fluorescence), extractable nuclear antigen detection by immunodiffusion (SSa, SSb, Scl70, RNP, Jo-1) and specific ELISA for anti-Jo1 antibodies. These patients were assigned to two groups: (1) group A which comprised 15 patients with acute onset of ILD (median duration between first symptom and diagnosis 5 weeks (range 0–8)) and acute respiratory failure needing oxygen therapy; and (2) group G which comprised 17 patients with a gradual onset of ILD (median duration between first symptom and diagnosis 26 weeks (range10–124)).
Analysis of respiratory and extrarespiratory manifestations according to initial presentation
Significant differences in general characteristics, respiratory manifestations and extrarespiratory findings between the two groups are shown in tables 1 and 2. One of the patients in group G had a history of Grave’s disease and another had a history of sarcoidosis (negative for anti-Jo-1 antibody at that time); one patient in group A was in remission from breast cancer; and three patients (one in group A and two in group G) were being treated with systemic corticosteroids and hydroxychloroquine or methotrexate for seropositive rheumatoid arthritis (negative for anti-Jo-1 antibody before the occurrence of ILD). There was no difference between the two groups in age at diagnosis or occupational exposure. We observed a high number of patients who had been exposed to cleaning products. Fever at admission and severe dyspnoea were more frequent in group A than in group G (table 2). In contrast, the proportion of cutaneous symptoms, arthralgia, myalgia and increased CK levels seemed to be similar in the two groups. EMG was performed in 28 patients with no differences in results between the two groups (table 2). Autoantibodies other than anti-Jo-1 antibodies were detected in nine patients in group G but in only one in group A (p = 0.007). All were negative for anti-Scl70 antibodies. Isolated rheumatoid factor was detected in three patients (one in group A and two in group G); isolated anti-Ro/SSa and La/SSb antibodies were detected in four patients in group G; and a combination of rheumatoid factor, anti-Ro/SSa and anti-La/SSb antibodies was detected in three patients in group G.
Functional parameters
Table 3 compares pulmonary function data in groups A and G. Three patients did not undergo functional testing on admission owing to the severity of ILD. In group A, median values of TLC, SVC, Tlco (expressed as percentages of normal predicted values) as well as arterial oxygen tension (Pao2) measured at rest in room air and expressed in mm Hg were significantly lower than in group G. No patient had an obstructive pattern and FEV1 did not differ between the two groups.
HRCT scan results
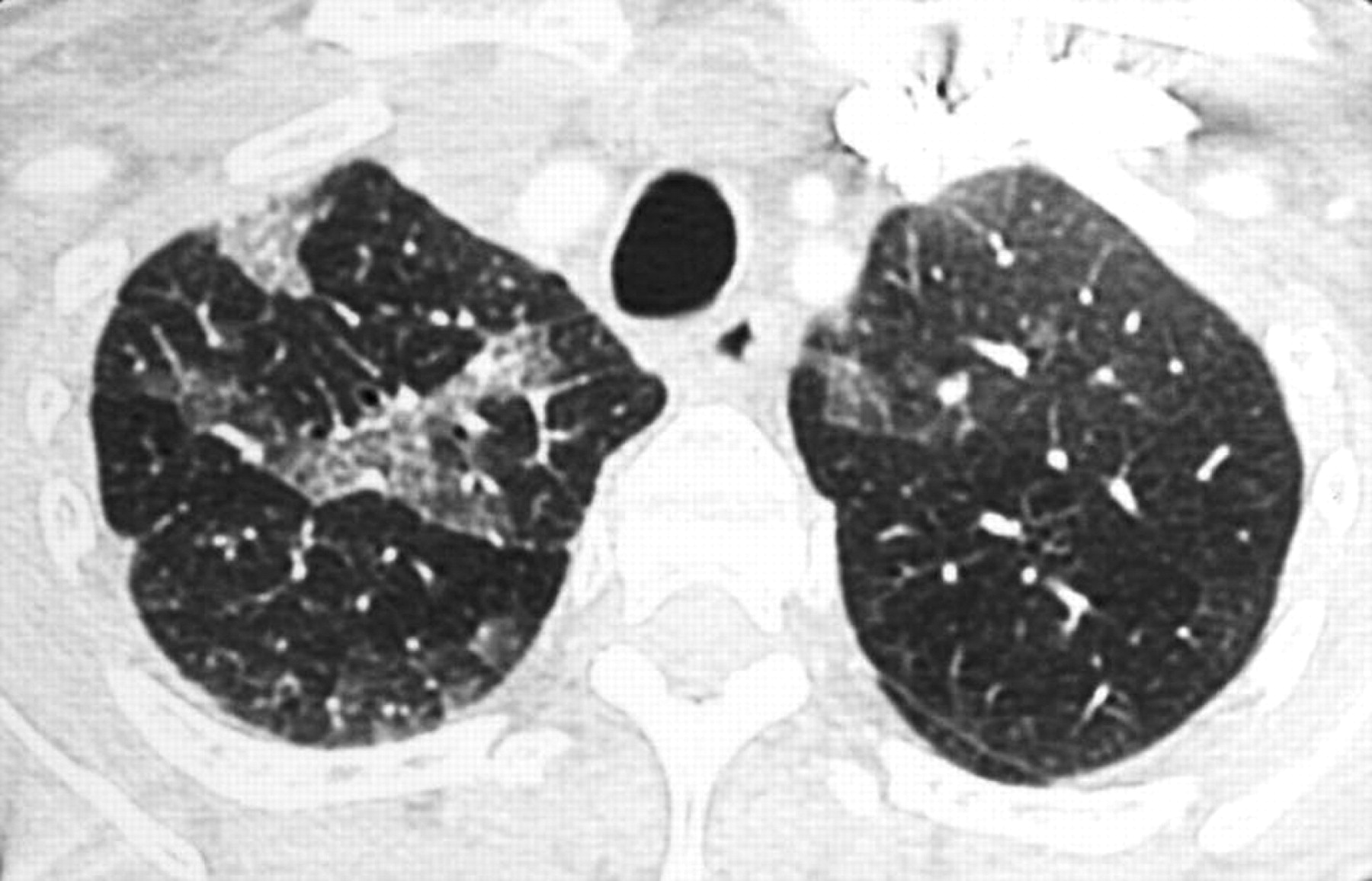
All patients underwent an HRCT scan. Table 4 shows the data obtained in the two groups. The predominantly inferior location was significantly more frequent in patients in group G, while consolidations were more frequent in group A. A combination of three patterns was observed in 80% of patients in group A: diffuse patchy ground-glass attenuation, basal predominance of irregular linear opacities and basal consolidations (figs 1 and 2). Honeycombing and traction bronchiectasis or bronchiolectasis were infrequent in both groups.


BAL and histological data
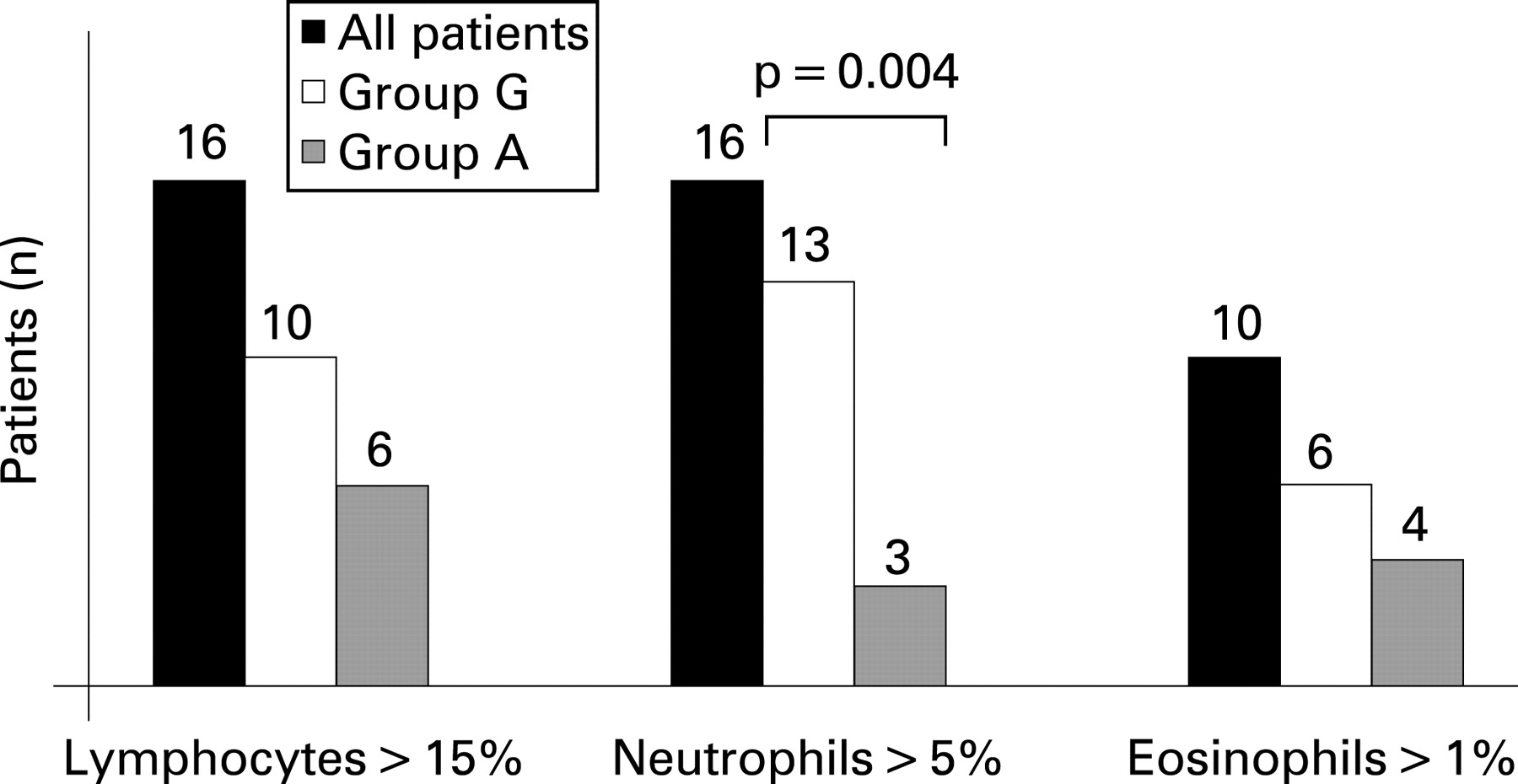
BAL results (expressed as total cells and percentages of lymphocytes, neutrophils, eosinophils and macrophages) were available for 25 of the 32 patients and are summarised in fig 3. The median number of total cells and the percentage of macrophages, lymphocytes, neutrophils and eosinophils did not differ between groups A and G. However, the proportion of patients with >5% of neutrophils in BAL fluid was higher in group G (13/2) than in group A (3/8) (p = 0.004). The CD4:CD8 ratio of BAL lymphocytes was <1 in all 12 cases (5 in group A and 7 in group G) evaluated for BAL T lymphocyte CD4 and CD8 phenotyping.
Histological data were obtained for 11 of the 32 patients (surgical lung biopsies (SB) in 6 cases and transbronchial biopsies (TBB) in 5 cases). Non-specific interstitial pneumonitis (NSIP) was diagnosed in 2 patients (2 SB; 1 in group A and 1 in group G) and suspected in 3 (3 TBB; 1 in group A and 2 in group G), cryptogenic organising pneumonitis (COP) was diagnosed in 2 patients (2 TBB; 2 in group A), usual interstitial pneumonitis (UIP) in 2 patients (2 SB; 2 in group G) and diffuse alveolar damage (DAD) associated with fibrosis and COP in 2 patients (2 SB; 2 in group A). Statistical analysis was not performed. We observed that DAD and COP were diagnosed only in group A.
Evaluation during the first year in groups A and G
At 3 months, two patients (6%) with in group A died on days 10 and 22 (the first was treated with corticosteroids only and the second with corticosteroids and cyclophosphamide; table 5). Both patients died from respiratory failure associated with DAD and, in one case, COP with fibrosis (necropsy). The percentage of patients showing improvement in ILD at 3 months was significantly higher in group A than in group G (13/15 vs 9/17; p = 0.004). In contrast, after 12 months (table 5), most patients in whom ILD continued to progress were in group A and were treated with corticosteroids alone. In group G, 88% of patients had stable ILD and most were on treatment with corticosteroids and immunosuppressive drugs (n = 10).
Evaluation at the end of the follow-up period
None of the 32 patients were lost to follow-up. The median duration of follow-up was 62 months (range 12–142) for the overall population, 41 months (range 12–112) in group A and 62 months (range 13–142) in group G. Twenty-five patients (82%) were still alive and six (18%) had died. Two patients died from respiratory failure with acute interstitial pneumonitis in the first month of follow-up (group A) and one died from severe sepsis attributable to immunosuppressive drugs (group G). Three patients died from neoplasia: one from brain metastasis related to primary lung adenocarcinoma (group A), one from ovarian cancer (group G) and one from non-Hodgkin lymphoma (group G).
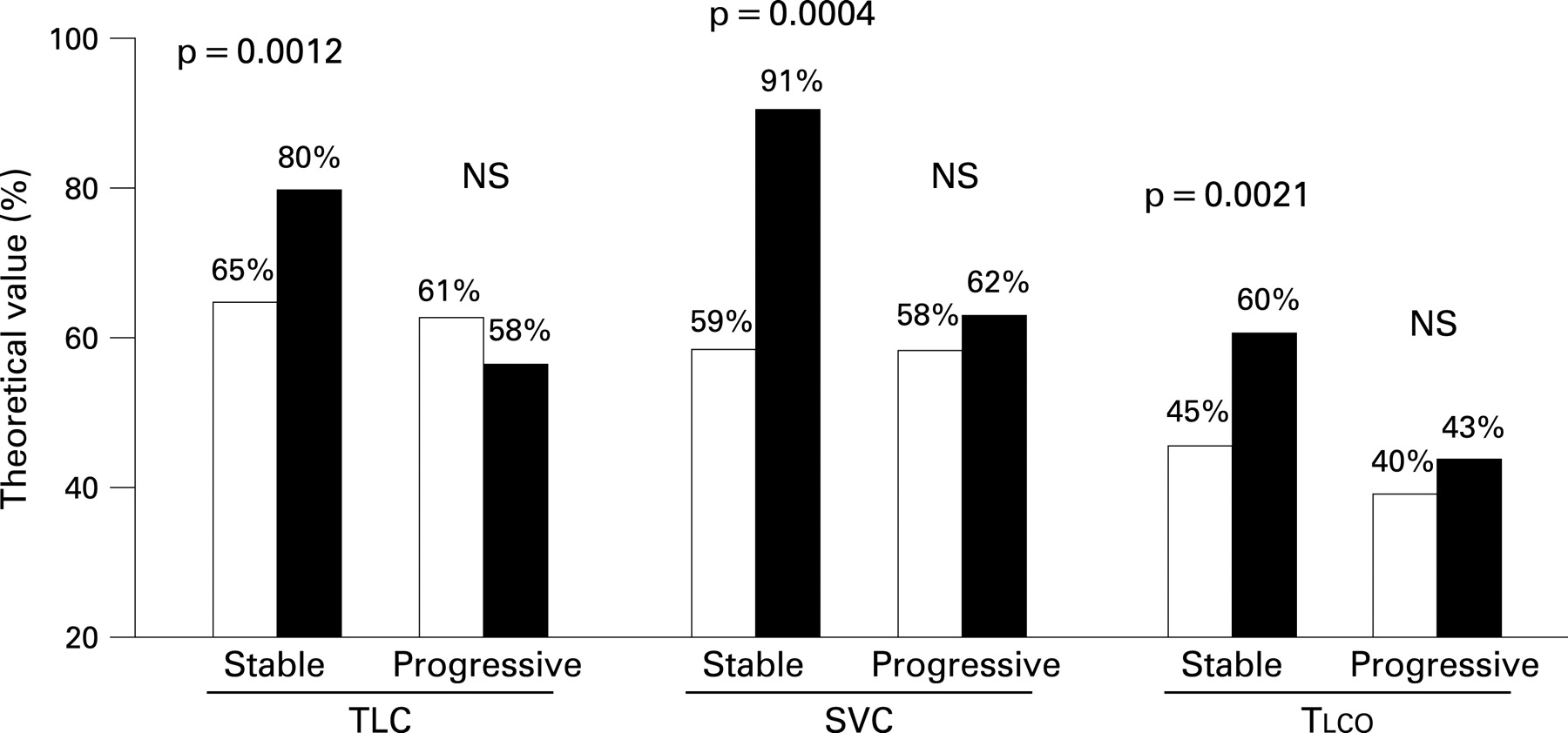
Among the 25 patients alive at the end of follow-up, 21 (84%) received immunosuppressants (cyclophosphamide in 8 patients, methotrexate in 4 and azathioprine in 9). ILD had remained stable in 17 of these 25 patients (based on dyspnoea, pulmonary function tests, Tlco, blood gas analysis and HRCT scan); 4 of these patients received corticosteroids alone, 12 were treated with corticosteroids and immunosuppressive drugs and one received immunosuppressive drugs alone. Eight patients displayed ILD progression with worsening of (or development of) honeycombing and traction bronchiectasis/bronchiolectasis on the HRCT scan and the need for long-term oxygen therapy; all eight were treated with a combination of corticosteroids and immunosuppressant drugs. Data comparing pulmonary function tests in patients with stable ILD and those with progressive ILD are shown in fig 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The characteristics on admission were compared between the 8 patients with ILD progression and the 17 with stable disease. The proportion of patients with gradual onset of presentation (2/8 vs 6/17), honeycombing on HRCT scan (4/8 and 5/17) and an increase in neutrophils in BAL fluid (1/5 vs 6/12) did not differ between those who progressed and those with stable disease. Furthermore, the proportion of patients with progressive ILD with myositis at presentation (3/8 vs 9/17) or during follow-up (5/8 and 13/17) was as frequent as that of patients with stable ILD. Finally, the initial treatment of patients with progressive ILD was corticosteroids in six patients and corticosteroids combined with immunosuppressants in two.
At the end of the follow-up period myositis was diagnosed in 18 patients (56%) (8 in group A and 10 in group G). It was associated with muscle weakness in 3 patients. In one case, nuclear magnetic resonance data linked myositis to corticosteroid treatment (group G). We failed to identify a relationship between the initial level of CK, the evolution of myositis and the outcome of ILD.
Adverse events associated with treatment
Seventeen patients (54%) experienced severe adverse effects linked to treatment (corticosteroids and/or immunosuppressants). Severe documented infections were reported in 11 cases and caused the death of one patient. Eight of the 11 severe infections concerned herpes zoster virus. No case of pneumocystosis was identified, although only five patients were given prophylactic sulphamethoxazole-trimethoprim treatment. Other adverse effects included organ failure, requiring hospitalisation in nine cases. There was no difference in adverse effects between groups A and G.
DISCUSSION
This study describes 32 patients who presented with initial ILD and anti-Jo-1 antibodies but who were not diagnosed as polymyositis or dermatomyositis at initial evaluation. This population of patients, who were referred to tertiary care centres for respiratory symptoms and who were treated for ILD according to international recommendations, was reasonably homogeneous, making it possible to evaluate the course of ILD and, in particular, the response to treatment under standard conditions. However, the study does have some limitations. Patients whose ILD developed before the possibility of detecting anti-Jo-1 antibodies, or patients with anti-Jo-1 antibodies and myositis treated before developing ILD, were not included in the study. However, patients with extrarespiratory symptoms such as myositis, arthralgia, Raynaud’s phenomenon or skin lesions were not excluded from the study.
In the anti-synthetase syndrome, respiratory involvement is associated with fever (87% cases), myositis (77–90%), arthritis (50–94%) and Raynaud’s phenomenon (62–93%).17 19 20 28 29 Although ILD is a recognised manifestation of the anti-synthetase syndrome, identified in 75–89% of cases in the principal studies,7 14 17 19–21 29 30 it remains poorly defined. Jo-1 antibodies directed against histidyl-tRNA synthetase account for about 80% of all cases of anti-synthetase syndrome12 and are not tissue- or disease-specific.16 31 32 However, there is a strong link between anti-Jo-1 antibodies and ILD: 75% of patients with ILD and polymyositis/dermatomyositis in the study by Schnabel et al had anti-Jo-1 antibodies.7
ILD may be associated with anti-Jo-1 antibodies as the sole clinical manifestation. Recent case reports have described ILD in patients with anti-Jo-1 antibodies without myositis.22–24 Santy et al24 reported NSIP in lung biopsies with CD8+ lymphocytic alveolar inflammation and anti-Jo-1 antibodies without myositis in 3 patients and Friedman et al22 identified 10 patients with anti-synthetase antibodies (anti-PL-12, Jo-1 and OJ) with ILD and no myositis. These two studies highlight the possible association of anti-synthetase antibodies with ILD, independently or as the sole manifestation of the anti-synthetase syndrome. The absence of myositis in these cases,22 24 and in 63% and 44% of patients at initial presentation and during follow-up in our study, requires further elucidation. In cases of isolated ILD associated with anti-synthetase antibodies, we cannot exclude the possibility of subclinical muscle involvement, as previously described.12 33 In some cases, myositis may have been prevented or controlled well enough by drugs to escape detection. In the absence of clinically or electromyographically apparent myositis, no muscle biopsy was performed. It was also difficult to identify myositis during the follow-up period when corticosteroid treatment may also have induced myopathy.
The study by Schmidt et al20 also confirms the diversity of the clinical manifestations associated with anti-Jo-1 antibodies. In 18 patients the clinical presentation was arthritis in 15 cases, myositis in 14 and ILD in 14. Only 4 patients had myositis at disease onset. One bias may result from the fact that patients with arthralgia and ILD were more likely to have prompted serological evaluation, including anti-Jo-1 antibodies, than patients presenting with isolated respiratory manifestations. Nevertheless, in the centres involved in the study, screening of patients for systemic disorders in cases of ILD was systematic at initial presentation. Indeed, it was noted that, upon admission, at least 9 patients (28%) had ILD without an increase in CK, myositis, arthralgia or cutaneous disorders.
Within the population studied, 32% were smokers, a proportion similar to the 30% observed in the French general population.34 Occupational exposure was identified in 39% of our patients and 26% of our population were employed in the cleaning industry. We cannot exclude the possibility that cleaning ladies exposed to abrasive agents may have developed pulmonary inflammation, as demonstrated in animal models.35 Exposure to toxic substances36 37 such as polymerising epoxy resin in Japan21 or silica36 has been shown to be associated with dermatomyositis and muscle weakness. Further studies are required to evaluate the impact of inhaled irritants, particularly in cleaning products, in order to identify a possible causal link between this occupation and ILD.
Our population also presented other connective tissue diseases and cancers. Overlap syndromes have been described in myositis.12 28 38 39 We identified patients with rheumatoid arthritis and Sjögren syndrome. None of our patients had systemic sclerosis. Sjögren syndrome is commonly associated with anti-Jo-1 antibodies, and anti-SSa antibodies are also associated with anti-Jo-1 without sicca syndrome.12 17 In our study, autoantibodies were more likely to be associated with the gradual onset of ILD. Cancer was identified during the follow-up period in three cases (lung, ovaries and non-Hodgkin lymphoma). Cancer is more frequent in cases of dermatomyositis than in the general population, and is slightly more frequent in cases of polymyositis,12 40 but no epidemiological association has been demonstrated between the anti-synthetase syndrome and cancer.
One particular feature of ILD associated with anti-Jo-1 antibodies was the high frequency of acute onset with fever and respiratory insufficiency reported in 47% of our patients on admission. Acute interstitial pneumonitis is a rare presentation of ILD, reported in dermatomyositis.30 41 Clawsson et al reported acute interstitial pneumonitis in three cases at initial presentation and during the course of anti-synthetase syndrome.42 ILD presenting with acute respiratory distress and fever may be due to infection (particularly pneumocystis), hypersensitivity pneumonitis (environmental or iatrogenic), alveolar haemorrhage and, on rare occasions, idiopathic “acute fibrosis” (initially described by Hamman-Rich).10 43 In cases of anti-synthetase syndrome with acute ILD, treatment with a high dose of corticosteroids (and other immunosuppressive drugs) is necessary to prevent rapid death due to respiratory insufficiency.42 44 We conclude that anti-Jo-1 antibodies should be sought and CK determined in cases of ILD with an acute presentation.
In 80% of patients with an acute presentation and in 35% of patients with gradual onset, the HRCT scan showed bilateral abnormalities with subpleural predominance, diffuse patchy ground-glass attenuation, basal consolidations and basal irregular linear opacities, possibly corresponding to NSIP in some patients.2 25 45 46 In the study by Marguerie et al,29 “pulmonary fibrosis” was identified in 15 of 19 patients with anti-Jo-1 anibodies; 16 of the 19 patients had myositis. “Fibrosis” was the term used to describe ILD and HRCT scans were not available at that time. Recent studies have described the pathology of the lung in myositis.1 2 8 10 42 47 ILD in polymyositis/dermatomyositis is identified principally as NSIP.1 2 10 In the acute presentation, DAD, fibrosis and COP have also been reported.2 8 10 42 UIP has only rarely been reported.2 8 10 45 In our series, lung biopsies in 5 of the 11 cases confirmed the frequency of NSIP. In the other cases, particularly those with an acute presentation, no lung biopsy was performed for ethical reasons.
The poorer prognosis for patients with anti-synthetase syndrome compared with those with myositis without autoantibodies has been found to be related to ILD.11 13 15 19 In myositis, Arsura et al11 found that 40–60% of patients with myositis and ILD died within 5 years. Data suggest that a combination of prednisolone and cyclophosphamide may improve the outcome in cryptogenic fibrosing alveolitis and lung fibrosis associated with connective tissue disease.48–50 The first-line treatment for ILD with Jo-1 antibodies12 51 is corticosteroids. In some cases an initial pulse of intravenous cyclophosphamide2 7 52 or a combination of a corticosteroid with azathioprine2 has been reported to induce remission. Santy et al showed that better responses were obtained if patients with Jo-1-positive ILD were treated with a combination of prednisone, azathioprine and cyclosporine.24 53 Nawata et al,54 Takizawa et al44 and Schnabel et al7 also suggested that ILD was frequently corticosteroid-resistant in patients with dermatomyositis/polymyositis. Schnabel et al7 found that disease progression was associated with increased CK levels at initial evaluation, which contrasted with the findings of Nawata et al54 who showed that patients with normal CK levels had a poorer prognosis. In our study we failed to identify a relationship between a poor outcome of ILD and the presence of myositis or an increase in CK levels. In the studies by Nawata et al and Schnabel et al, patients were diagnosed as dermatomyositis or polymyositis before ILD and they did not focus on anti-Jo-1 antibodies.7 54
Schnabel et al differentiated two forms of ILD associated with polymyositis/dermatomyositis: one with a progressive and the other with a non-progressive course.7 Based on an evaluation of 10 patients with progressive ILD, he concluded that disease activity should be evaluated based on the presence of ground-glass opacities on the HRCT scan and neutrophils in the BAL fluid. In those patients, an intravenous pulse of cyclophosphamide in combination with prednisolone seemed be more effective than corticosteroid alone.7 In our study, 87% of patients in group A and 53% of patients in group G showed improvements in ILD at 3 months. Most of the patients in the two groups were treated with corticosteroid alone. Some of the patients in group A suffered respiratory failure after 1 year; most of those patients had been treated with corticosteroid alone. Thus, ILD, particularly with an acute presentation, responds to corticosteroids but relapses are not completely prevented by corticosteroid alone. In group G, fewer relapses were observed during the first year but most of the patients had already been treated with a combination of corticosteroids and immunosuppressants. At the end of the follow-up period, ILD was stable in 68% of our patients, with no difference between the two groups. However, by that time, most patients had been treated with a combination of corticosteroids and immunosuppressants. The outcome was poor in about one-third of patients who suffered severe respiratory insufficiency. At the end of the study a few patients remained stable on corticosteroid alone.
In conclusion, ILD may be associated with anti-Jo-1 antibodies as the sole clinically apparent manifestation and is characterised by a high frequency of acute onset associated with respiratory insufficiency, fever and specific HRCT scan patterns. The immediate response to corticosteroid alone was good, but recurrences frequently occurred. At the end of the study about two-thirds of patients had stable ILD, while the other third displayed progression with respiratory insufficiency. A combination of corticosteroid and immunosuppressant drugs was required to control ILD in most cases. Severe adverse effects of treatment were observed and infection with varicella zoster virus was frequent. Early testing for anti-synthetase antibodies, particularly anti-Jo-1, and CK determination are useful investigations in patients presenting with ILD, particularly when the latter is associated with acute onset, fever, basal consolidations, basal irregular lines and diffuse patchy ground-glass opacities on the HRCT scan; 47% of the patients included in this study presented with such a combination of signs. A prospective evaluation of patients with ILD is required to establish the true frequency of anti-Jo-1 antibodies in “idiopathic” ILD.
REFERENCES
Footnotes
Competing interests: None.