Article Text

Abstract
Cystic lung disease is a frequently encountered problem caused by a diverse group of diseases. Distinguishing true cystic lung disease from other entities, such as cavitary lung disease and emphysema, is important given the differing prognostic implications. In this paper the features of the cystic lung diseases are reviewed and contrasted with their mimics, and the clinical and radiographic features of both diffuse (pulmonary Langerhans’ cell histiocytosis and lymphangioleiomyomatosis) and focal or multifocal cystic lung disease are discussed.
- AML, angiomyolipoma
- CCAM, congenital cystic adenomatoid malformation
- DIP, desquamative interstitial pneumonia
- HRCT, high resolution computed tomography
- LAM, lymphangioleiomyomatosis
- LIP, lymphocytic interstitial pneumonia
- PLCH, pulmonary Langerhans’ cell histiocytosis
- TSC, tuberous sclerosis complex
Statistics from Altmetric.com
- AML, angiomyolipoma
- CCAM, congenital cystic adenomatoid malformation
- DIP, desquamative interstitial pneumonia
- HRCT, high resolution computed tomography
- LAM, lymphangioleiomyomatosis
- LIP, lymphocytic interstitial pneumonia
- PLCH, pulmonary Langerhans’ cell histiocytosis
- TSC, tuberous sclerosis complex
The identification of air-containing lucencies within the pulmonary parenchyma on radiographic studies is a frequently encountered problem by all those who care for patients with lung disease. The availability of high-resolution computed tomographic (HRCT) scanning, with both its sensitivity and specificity for defining abnormalities of the lung parenchyma, has dramatically improved our ability to characterise these pulmonary lesions. They can be usefully classified as either cysts or cavities, with cysts including subcategories of bullae, blebs and pneumatoceles. While all refer to an air-filled lucency within the lung parenchyma, each denotes a specific radiographic feature that can be differentiated on the basis of wall thickness, size, overall number and anatomical distribution. Why make distinctions among these radiographic features and their patterns? The differential diagnosis possibilities associated with each pattern may differ substantially, such that the evaluation and subsequent management may be fundamentally altered.
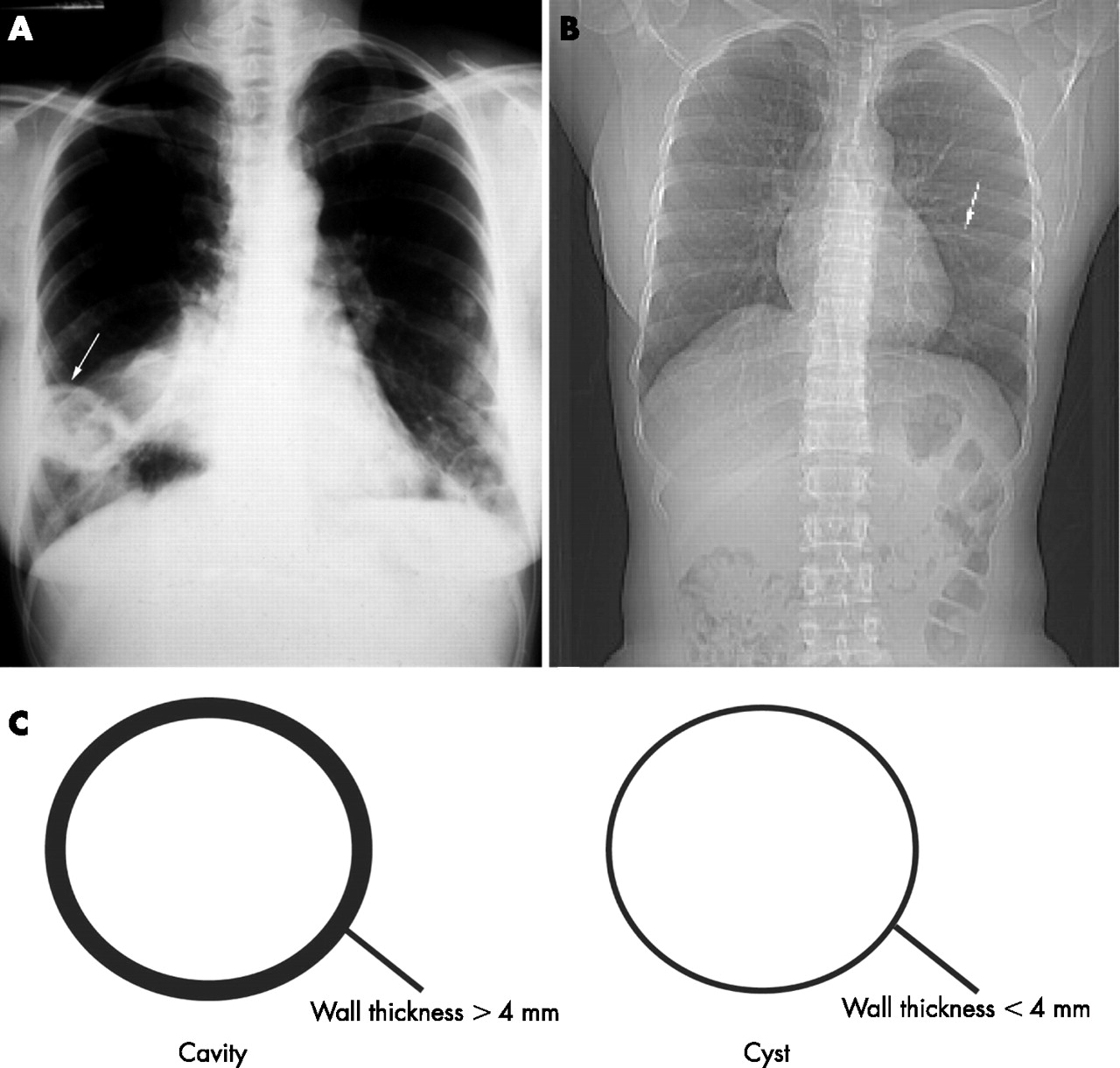
Cysts refer to air-filled spaces with sharply demarcated thin walls (<4 mm) whereas cavities refer to air-filled lesions with thick walls (>4 mm) (fig 1).1 A bulla is a cyst >1 cm in diameter with a smooth wall <1 mm in thickness.1 In contrast, blebs are cysts usually <1 cm in diameter, subpleural or pleural in location, and typically in an apical distribution.1,2Pneumatoceles are cysts which accompany primary pulmonary infections or chest trauma and frequently resolve with treatment of the associated infection.

Posteroanterior chest radiographs demonstrating a cavitary lesion (arrow) in Wegener’s granulomatosis (A). In contrast, note the thin-walled cyst (arrow) in a patient with Sjögren’s disease (B). Cavities are defined as air-filled lesions with thick walls >4 mm whereas cysts are sharply demarcated thin walls <4 mm (C).
Cysts and cavities must be differentiated from a number of other radiographic features that may mimic them including emphysema, loculated pneumothoraces, honeycomb lung and bronchiectasis (both traction and suppurative). Emphysema differs from cystic lung disease as the term refers to irregular asymmetrical areas of decreased lung attenuation and decreased vascularity (termed “arterial deficiency”) which do not have a defined wall (fig 2).3,4 Honeycombing is defined as a cluster or row of cysts that are usually <5 mm in diameter and associated with end-stage lung fibrosis. Honeycombing is most often subpleural in distribution and accompanied by other features of lung fibrosis such as reticulation and traction bronchiectasis (fig 3).5 Honeycombing may be seen as a result of any disease that results in pulmonary fibrosis. Bronchiectasis, or the dilatation and distortion of bronchi and bronchioles, may be mistaken for cystic airspace disease when a dilated airway is viewed “en face” (fig 3). Bronchiectasis may be the result of either a chronic suppurative process or accompany lung fibrosis, when it is then referred to as traction bronchiectasis. In traction bronchiectasis, bronchi and bronchioles are dilated or stented open as a consequence of increased elastic recoil.6 They can be differentiated from cystic lung disease by the presence of an adjacent blood vessel suggesting a bronchovascular unit rather than a cystic air space. Lastly, a loculated pneumothorax may mimic a cystic air space disease, but can be differentiated from true cysts based on its distribution, failure to adhere to a defined anatomical unit and relationship to the pleural surface.

High-resolution CT scan showing emphysematous air space disease (arrow). In contrast to cystic lung disease, discrete walls are not present.

High-resolution CT scans from two patients with idiopathic pulmonary fibrosis showing traction bronchiectasis (white arrows). The adjacent bronchi and vasculature differentiates these structures from true cysts. End-stage lung fibrosis is evident with diffuse honeycombing (black arrow), reticulation and traction bronchiectasis.
In this paper we review the aetiology, clinical and radiographic characteristics of congenital and acquired cystic lung disease, with a particular focus on two diffuse diseases (pulmonary Langerhans’ cell histiocytosis (PLCH) and lymph-angioleiomyomatosis (LAM), and two focal or multifocal cystic lung diseases (desquamative interstitial pneumonia (DIP) and lymphocytic interstitial pneumonia (LIP)). We will only briefly review cavitary lung disease, primarily as it contrasts with cystic lung disease, and refer the reader to a recent article by Ryu and Swensen for an in-depth review of this topic.7
CAVITARY LUNG DISEASE
Cavities are thick-walled (>4 mm) intraparen-chymal airspace opacities whose causes are numerous and include congenital, infectious, inflammatory/autoimmune and neoplastic dis-orders (table 1). Because of prognostic and therapeutic implications of the underlying diagnosis, a definitive evaluation is required.
Aetiology of cavitary lung disease
Of paramount importance is determining whether a lesion is neoplastic, and the thickness of the cavity wall is useful in this regard. Solitary cysts (wall thickness <4 mm) are benign in more than 90% of cases.8 In contrast, solitary cavities with a wall thickness >15 mm are malignant 95% of the time.8 Cavities with walls >5 mm but <15 mm are equally as likely to be malignant as they are benign. Overall, the majority of solitary cavities are neoplastic and represent bronchogenic carcinomas (most commonly squamous cell carcinoma), although rarely pulmonary lymphomas can cavitate.9
Chronic cavities, those present for >1 month, are more likely to be malignant, congenital or the result of chronic inflammatory disorders.7 Acute or subacute cavities, those present for <1 month and accompanied by the recent development of signs and symptoms, are often due to infections, inflammatory disorders (such as Wegener’s granulomatosis), thromboemboli/septic emboli or trauma (fig 4). Infectious cavities may occur during or subsequent to a necrotising pneumonia (Staphylococcus aureus or Streptococcus pneumonia), post-primary tuberculosis (often with a posterior upper lobe preference) or chronic infection with opportunistic or endemic fungi (nocardiosis, sporotrichosis, aspergillosis, cryptococcosis, mucor-mycosis, histoplasmosis, blastomycosis or coccidioidomycosis). Pulmonary actinomycosis, which can mimic a bronchogenic carcinoma, may present as a persistent pulmonary mass or consolidation with cavitation.10 Atypical causes of cavities, which are not frequently seen outside endemic areas, include paragonimiasis, melioidosis and echinococcosis and should be considered depending on the clinical scenario.

Posteroanterior radiograph showing a lung cyst in the left mid-lung zone following blunt chest trauma.
CYSTIC LUNG DISEASE
Cystic lung disease may be subclassified into two types: (1) discrete, focal or multifocal cysts and (2) diffuse cysts with a pan-lobular distribution. Several congenital and acquired diseases may produce focal or multifocal cystic lung disease (table 2).
Aetiology of cystic lung disease
Focal or multifocal cystic lung disease
Congenital abnormalities
Bronchogenic cysts, pulmonary sequestration and congenital cystic adenomatoid malformations (CCAM) are congenital disorders that present clinically in early childhood or young adulthood. Bronchogenic cysts represent congenital abnormalities resulting from abnormal budding of the developing tracheobronchial tree. While they typically appear as either mediastinal or intraparenchymal homogeneous masses, they may also manifest as thin-walled cystic cavities with or without an air-fluid level, pneumothoraces or recurrent pneumonia. They do not communicate with the tracheobronchial tree unless they become secondarily infected, which occurs in >75% of intraparenchymal cysts. Once infected and in communication with the tracheobronchial tree, the cysts often rapidly enlarge, some to >25 cm, due to a check valve mechanism. Haemoptysis often develops when this occurs. Mediastinal bronchogenic cysts may present with dyspnoea, stridor, cough or chest pain, depending upon the location, secondary to a mass effect and compression from the lesion.
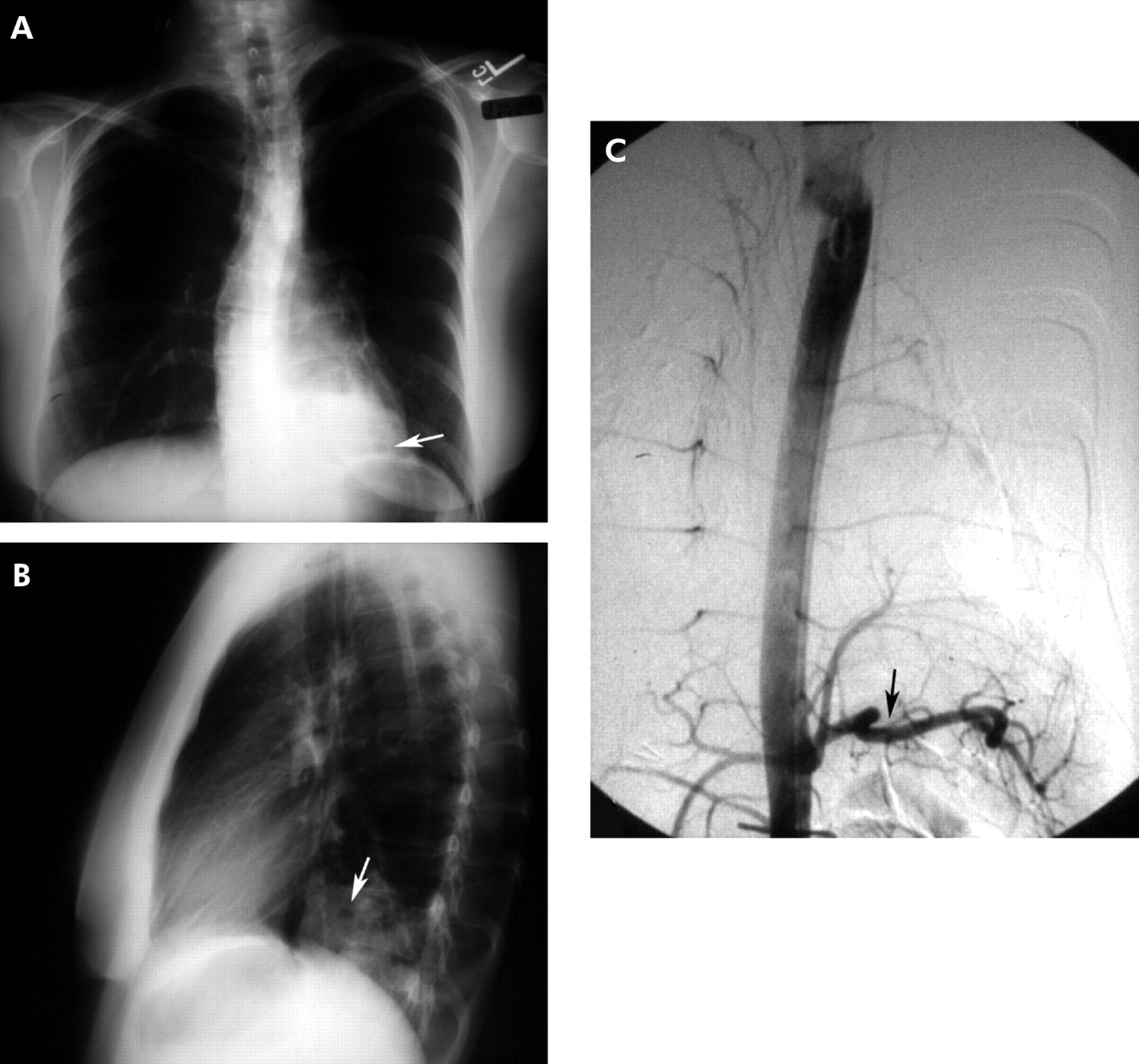
Bronchopulmonary sequestration is one of the more common congenital pulmonary malformations. In sequestration, a portion of lung develops with its own systemic arterial blood supply isolated from the main body of the lung. Two variants exist: intralobar sequestration (encompassed by the same visceral pleural envelope as the adjacent surrounding lung) and extralobar sequestration (encompassed by a separate distinct visceral pleura); 75% of sequestrations are intralobar with two-thirds of them occurring in the posterior basilar segment of the left lower lobe and up to one-third in a similar location on the right.11 Upper lobe intralobar sequestration is rare but, if present, may be accompanied by other congenital malformations. Most sequestrations are diagnosed in childhood or in young adults presenting with a history of recurrent pneumonia. In the adult the age at the time of diagnosis ranges from 20 to 70 years.12 As in intralobar sequestration, extralobar sequestration occurs most often on the left and is often peripheral and presents as a wedge-shaped density associated with the left diaphragm in 90% of cases. While the majority of sequestrations present radiographically as homogeneous masses, cysts may also be seen (fig 5). A definitive diagnosis is made by demonstrating its systemic arterial blood supply.

(A) Posteroanterior and (B) lateral radiographs showing cystic airspace abnormalities in the left lower lobe, posterior segment. An intralobar sequestration is confirmed by angiography which shows a systemic arterial supply arising from the aorta (C).
CCAM refers to a group of pulmonary anomalies believed to represent localised arrested development of the fetal bronchial tree. Most cases are identified with prenatal ultrasonography.13 In those not identified antenatally, patients are usually diagnosed within 1 month of birth. Some individuals, however, have been diagnosed as late as 60 years of age.14 Three subtypes of CCAM exist: type I (cystic), type II (intermediate) and type III (solid). Type I cystic adenomatoid malformations are the most common and represent more than half of all CCAMs.14 In the newborn infant, cystic and intermediate CCAMs begin as homogenous masses that become air filled in the days to weeks following birth. CT scanning is superior to plain chest radiography and shows multiple thin-walled complex cystic masses ranging in size from 2 to 12 cm. The cysts may contain air, fluid or both. CCAMs may occur in either the upper or lower lobes with up to 15% of cases being bilateral.15–17 Most patients will present with neonatal respiratory distress. Pneumothorax or recurrent pneumonia are less common but represent the typical manifestation in adults.16 Bronchoalveolar carcinoma can complicate cystic CCAMs.18 In those that present after birth, CT imaging is the diagnostic entity of choice. Surgical resection, preferably a lobectomy, is curative. Recurrences have been described in patients in whom more selective resection was attempted, presumably due to residual anomalous tissue.19
Infections
Several infections may present with pneumatoceles, a specific subcategory of cystic airspace disease. Pneumatoceles are cysts that present acutely or subacutely and are progressive until the underlying infection is treated, after which they usually resolve. They occur most commonly with Staphylococcus aureus and Pneumocystis carinii pneumonias and are more common in children. True infectious cysts that persist despite resolution of the primary infection may occur with Pneumocystis carinii pneumonia or echinococcosis (hydatid disease). Hydatid cysts occur as a result of infection with Echinococcus granulosus or E multiloculus and are often multiple, occurring most commonly in the upper lobes. The cysts associated with Pneumocystis carinii pneumonia most often occur in patients with advanced HIV infection and AIDS, are multiple, usually apical in distribution, and range in size from 1 to 5 cm in diameter. Although isolated cysts may occur in coccidioido-mycosis, cavitary disease from a necrotising infection remains the more common manifestation of this infection.
Lymphocytic interstitial pneumonia
Lymphocytic interstitial pneumonitis (LIP) is a rare interstitial lung disease which was originally described by Carrington and Liebow in 1966.20 Both idiopathic and secondary forms are described, many of which are associated with some form of dysproteinaemia or autoimmunity.21–26 50–77% of patients with LIP, including those with the idiopathic variety, have some associated serum protein abnormality, most often polyclonal gammopathy (IgG or IgM)27–33 but also hypogammaglobulinaemia.27,34 Sjögren’s syndrome accounts for at least 25% of reported cases.21,29,31,35–37 Several cases of common variable hypogammaglobulinaemia have been complicated by LIP29 with a high incidence of conversion to a lymphoreticular malignancy.38 Retroviral (HIV-1, HTLV-1) and Epstein-Barr viral infections have been identified in some patients with LIP, but their exact role remains unclear.21,26,39–42
LIP occurs more commonly in women and presents between the fourth and seventh decades of life with a mean age at diagnosis of 56 years.29,30,43,44 It may occur in children, particularly those with hypogammaglobulinaemia or AIDS,45–48 and a familial form of LIP has also been reported.49,50 The most common presenting symptoms are progressive dyspnoea and cough.28,29,36 Additional symptoms include weight loss, pleuritic pain, arthralgias and fever. Clubbing and bibasilar rales are common physical findings. It is distinguishable from other interstitial pneumonias by the presence of monotonous sheets of lymphocytes expanding the interstitium and compressing alveolar spaces.27 Alveolar collections of lymphocytes as well as perilymphatic and perivascular aggregates may also occur in LIP.51,52 This lymphocytic infiltrate is associated with type II cell hyperplasia; the accumulation of large interstitial reticuloendothelial cells, mononuclear cells and giant cells forming non-caseating granulomas;21,53 perivascular and paraseptal amyloid deposition;37,44,45,53,54 and well-formed lymphoid germinal centres.20 The lymphocyte aggregates in LIP are polyclonal; the few reports of monoclonal lymphocyte populations raise the concern of a premalignant condition.55–57 B cells and T cells are both present, with B cells typically located in the lymphoid nodules whereas T cells are primarily located in the lung interstitium.27,35,44
Radiographically, bibasilar reticulonodular infiltrates are common, with single or multiple well-circumscribed masses often coalescent with air bronchograms. However, prominent cysts occur in up to two-thirds of patients.58 The thin-walled cysts are multifocal and variable in size and shape. A series of CT scans of 14 patients with LIP followed over a median of 13 months reported ground glass attenuation, thickening of interlobular septae, focal consolidation, centrilobular nodules and cystic airspaces (fig 6). All the parenchymal abnormalities were reversible with treatment with the exception of the cysts, and new cysts developed in areas of prior centrilobular nodularity.59 Other CT features that have been reported include lymph node enlargement (in 68% of patients), subpleural small nodules (in 86%) and thickening of bronchovascular bundles (in 86%).60 Mixed alveolar-interstitial and a micronodular pattern, honeycombing as well as signs of pulmonary hypertension have been reported.30,54,61,62 Rarely, recurrent pneumo-thoraces may be a presenting sign.63 Pleural effusions are rare, except in AIDS-related LIP, and should raise the concern for a lymphoproliferative disorder.62

(A) Posteroanterior radiograph showing diffuse reticulonodular infiltrates with associated cysts (arrow) in lymphocytic interstitial pneumonia. The cysts are peribronchiolar in distribution (arrow) based on high-resolution CT scans (B).
The prognosis in LIP is variable with four potential outcomes: (1) resolution following corticosteroid therapy alone or in combination with other immunosuppressive drugs;30,35 (2) progressive pulmonary fibrosis;29,30 (3) overwhelming pulmonary or systemic infection;29,30,34 and (4) conversion to lymphoma.27,30,54,64 Median survival is reported as 11.5 years.65 The data regarding response to treatment are uncontrolled, often unaccompanied by objective testing, with dosage schedules differing among reports. However, progressive disease despite aggressive treatment can occur. Unfortunately, there are no clinical, laboratory or histological parameters that help to predict an individual’s outcome. Bacterial superinfection is a common cause of death, especially in those with hypogammaglobulinaemia.29,30,34
Desquamative interstitial pneumonia
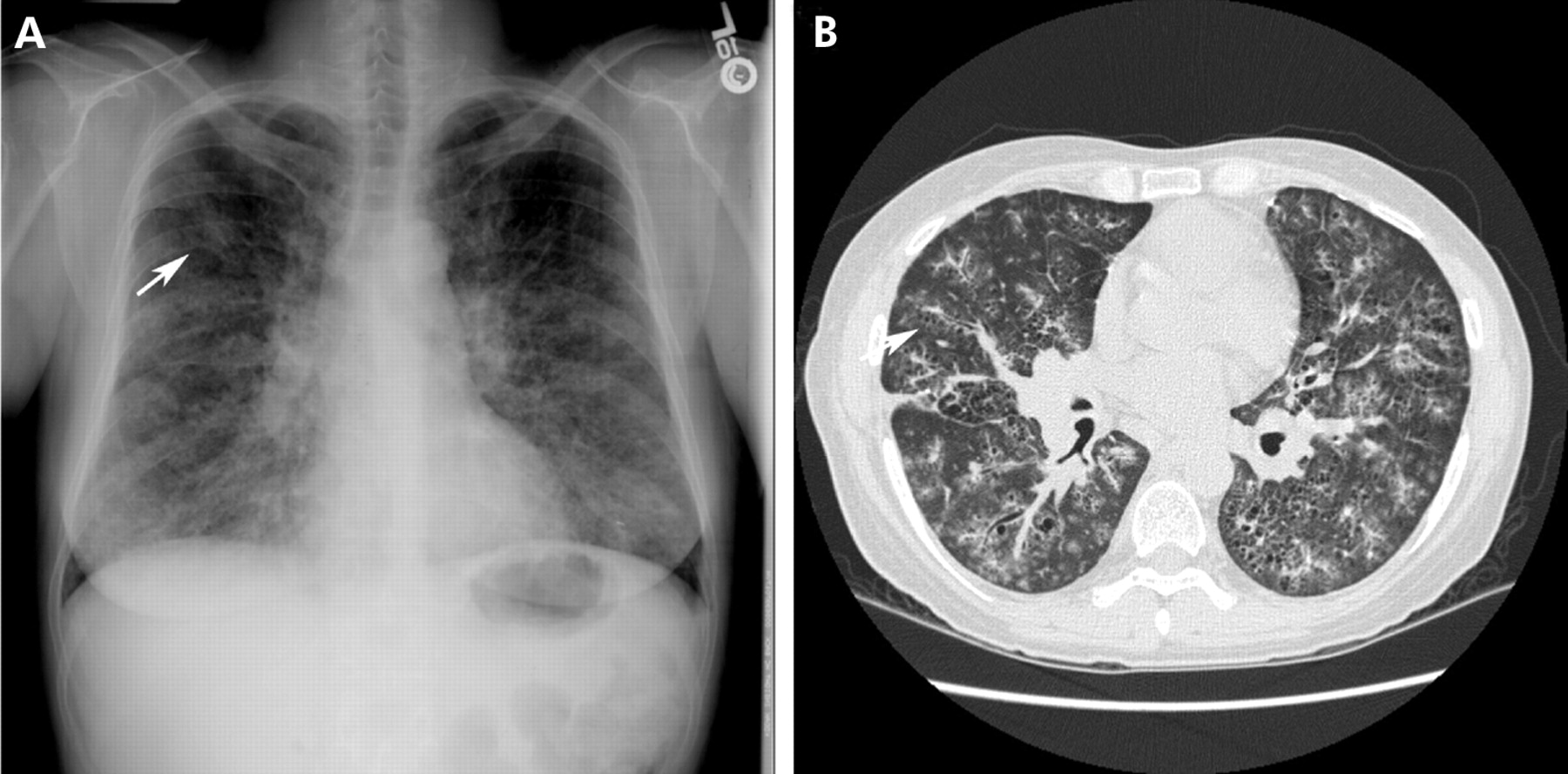
Desquamative interstitial pneumonitis (DIP) is a rare idiopathic interstitial pneumonia first described by Liebow.66 It is a disease that is seen almost exclusively in current or former smokers. Men appear to be affected twice as often as women and the mean age of onset is 42 years.67 Cough and dyspnoea are the most common symptoms, occurring subacutely over weeks to months.68 Pathologically it is characterised by the accumulation of pigmented macrophages within the airspaces with a homogenous appearance and limited mononuclear infiltrate within the interstitium.67,69,70 This contrasts with the peribronchiolar accumulation of macrophages in respiratory bronchiolitis interstitial lung disease (RB-ILD). Its characteristic radiographic manifestations include diffuse ground glass opacities with parenchymal cysts (fig 7). In 25–44% of patients a diffuse ground glass pattern is seen in the middle to lower lung zones on plain chest radiographs and in all patients on HRCT imaging.71 Cysts were present in six of eight patients in one series and appeared to resolve following immunosuppressive therapy.68,72 In contrast to honeycomb cysts, which may also occur in DIP, isolated parenchymal cysts occur within areas of ground glass, remote from areas of end-stage fibrosis and honeycomb cystic disease.68

(A) Posteroanterior radiograph from a patient with desquamative interstitial pneumonitis showing diffuse ground glass opacities with cysts (arrow). (B) A high-resolution CT scan from the same patient more clearly shows cysts within ground glass opacities. Traction bronchiectasis is also present, suggesting that the ground glass opacities are secondary to fibrosis rather than an inflammatory process.
Treatment for DIP consists of tobacco cessation and corticosteroids. More than two-thirds of patients symptomatically and radiographically improve with corticosteroids. However, progressive deterioration despite treatment may occur in up to 30% of patients.73 Overall survival is significantly better than that experienced by patients with idiopathic pulmonary fibrosis (IPF), approaching 70–100% at 10 years.74,75
Diffuse cystic lung disease
Diffuse cystic lung disease is invariably one of two rare disorders: pulmonary Langerhans’ cell histiocytosis (PLCH) or lymphangioleiomyomatosis (LAM).
Pulmonary Langerhans’ cell histiocytosis
Pulmonary Langerhans’ cell histiocytosis (PLCH) is a rare interstitial lung disease seen primarily in young adults. While the terms eosinophilic granulomatosis and pulmonary histiocytosis X have previously been used for this disorder, the currently preferred term is PLCH. It is part of a spectrum of systemic diseases termed Langerhans’ cell histiocytosis. In these disorders the lung, bone, pituitary gland, thyroid, skin, lymph nodes and liver may be involved in isolation or combination. There is an established classification system to denote the various forms of isolated or multiorgan involvement.76 PLCH indicates clinically significant lung involvement with or without additional organ involvement.
PLCH affects individuals in their 20s and 30s and, similar to DIP, the vast majority are active tobacco smokers.77,78 Patients typically present with non-productive cough and dyspnoea, occasionally accompanied by chest pain which is often pleuritic.79 Pneumothorax is the initial manifestation in 15% of patients and commonly recurs.80,81 Haemoptysis is less common, occurring in up to 5% of patients. Fever, anorexia and weight loss may occur in up to one-third of patients. Approximately 25% are asymptomatic at the time of presentation.82
Langerhans’ cell infiltration is the pathological hallmark. These cells are derived from monocyte-macrophage progenitors and function as antigen presenting cells. The presence of Birbeck granules or pentalaminar rod-shaped intracellular structures on electron microscopy and CD1a immunoreactivity make these cells unique.83 Eosinophils, lymphocytes, fibroblasts and multinucleated giant cells accompany Langerhans’ inflammation. The infiltration and subsequent Langerhans’ cell monoclonal proliferation occurs in a bronchiolocentric distribution with micronodule formation (1–5 mm). As the disease progresses, nodules evolve from a predominantly cellular lesion to a fibrotic one, with a characteristic stellate appearance. Destruction of the bronchiolar wall leads to bronchiolar dilation and cyst formation. Histopathological-radiographic correlation suggests that the cellular bronchiolocentric nodules correspond to radiographic nodules whereas cavitary nodules and fibrous cysts correspond to cysts.
Radiographic studies reveal a characteristic combination of diffuse cysts and centrilobular micronodules (fig 8).84 In an active smoker the combination is virtually diagnostic. Early in the course of the disease nodules may dominate whereas, over time, diffuse cysts become more common. The abnormalities are diffuse and symmetrical with a characteristic sparing of the costophrenic sulci. The cysts in PLCH vary in size and shape, in contrast to the uniform appearance of cysts in lymphangioleiomyomatosis (LAM).85,86 Most cysts are <10 mm in diameter but, with progression, confluence of adjacent cysts occurs, leading to irregular-shaped cystic airspaces, enlarging up to 80 mm in diameter.87 In contrast to most interstitial lung diseases, lung volumes may be preserved in PLCH.80 In one series of 81 patients pulmonary physiology was normal in 13.6% of patients. Obstructive (27.2%), restrictive (45.7%) or mixed defects (4.9%) are commonly seen.88 Reduced carbon monoxide transfer factor is present in 60–90% of patients.77 Dilation of the main and central arteries may occur over time, suggesting the development of pulmonary hypertension89 which may be due to vascular involvement with intimal fibrosis, medial hypertrophy or luminal obliteration.90,91

(A) Posteroanterior radiograph from a patient with pulmonary Langerhans’ cell histiocytosis (PLCH). Diffuse reticulonodular infiltrates are present with sparing of the costophrenic sulci (arrow).(B) High-resolution CT scan showing diffuse irregularly shaped cysts (white arrow) with adjacent nodules (black arrow), features that characterise PLCH.
PLCH may evolve in one of three ways: progression, stabilisation or resolution. Smoking cessation is the cornerstone of treatment, but this alone does not guarantee resolution or even stabilisation. However, most patients experience some degree of stabilisation in the short term. Corticosteroids may be beneficial, although there limited data are available to support their use. It is reasonable to consider a trial of corticosteroid treatment in patients who have progressive disease despite smoking cessation. In our experience, those patients with early primarily nodular infiltrates appear to have the most beneficial response to corticosteroids. In patients who develop pneumo-thoraces, early pleurodesis reduces the risk of recurrence and is not a contraindication for subsequent lung transplantation.81
The prognosis for most patients with PLCH is good with 5 year and 10 year survival rates of 74% and 64%, respectively.88 The estimated mean survival from the time of diagnosis is 13 years.88,92 In two retrospective studies, 27% of patients died or required transplantation for progressive disease. Recurrence within the donor lung has been reported by two separate groups.93,94
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare diffuse lung disease that is characterised by an abnormal proliferation of atypical smooth muscle cells within the lung, kidney, lymphatics or any combination.95 It may occur sporadically or in association with the neurocutaneous syndrome, tuberous sclerosis. Approximately 400 cases of LAM have been reported, most of which were sporadic.96 Recent studies suggest that up to one-third of women with tuberous sclerosis have a subclinical cystic lung disease consistent with LAM.96 The peak prevalence is in the third and fourth decade, primarily in women of child-bearing age but post-menopausal women have been diagnosed with LAM.97,98 In contrast to PLCH, there is no association with smoking.
Patients with either sporadic or the tuberous sclerosis-associated form may present with pulmonary or extrapulmonary signs and symptoms. Progressive dyspnoea and pneumo-thorax are the two most common initial presentations in sporadic LAM.98,99 Dyspnoea may be due to either diffuse cystic lung disease or secondary to a chylous effusion, a manifestation of an obstructing lymphangiomyoma.100 Pulmonary function tests are abnormal in most patients, with a decreased carbon monoxide transfer factor occurring most frequently.99 Hypoxaemia, obstruction with an increased residual volume and mixed obstructive-restrictive defects may also be seen. Up to 26% of patients may have improved airflows after bronchodilator therapy.99 Extrapulmonary manifestations are common at presentation, particularly in the abdomen. The most common intra-abdominal findings include renal angiomyolipomas (AML), enlarged abdominal lymph nodes and lymphangiomyomas (cystic spaces filled with low attenuation fluid) in 10–20%. Ascites, dilation of the thoracic duct and hepatic AML can also be seen.101 Flank pain, haematuria and, rarely, abdominal haemorrhage may occur secondary to an AML which occur in 30–50% of patients with sporadic LAM and 70% of patients with tuberous sclerosis-associated LAM and range in size from 0.5 to 20 cm.96,102,103 Large screening programmes in patients with tuberous sclerosis have shown that patients may also present with an abnormal radiograph without significant pulmonary symptoms.104,105
Abnormal smooth muscle proliferation in LAM has been linked to the tuberous sclerosis complex (TSC) as mutations in TSC1 and TSC2, the genes encoding hamartin and tuberin, have been identified in patients with tuberous sclerosis-associated LAM and sporadic LAM. In tuberous sclerosis, germline mutations in TSC1 are heterozygous. LAM occurs in these patients following loss of heterozygosity of TSC1. In sporadic LAM, homozygous somatic mutations in TSC2 are present in the lung lesions and angiomyolipomas of patients.106 Following the loss of tuberin, a failure of signalling through the TSC pathway occurs, leading to dysregulated endocytosis, cellular proliferation, and loss of control of cell cycle with resulting abnormal smooth proliferation.107,108
Common to pulmonary and extrapulmonary LAM are foci of abnormal smooth muscle cells termed LAM nodules. Two distinct populations of cells exist: small spindle-shaped cells which are located at the centre of LAM lesions and large epithelioid cells abutting the periphery. Each cell has a different rate of proliferation and can be differentiated based on its immunoreactivity with HMB45, a pre-melansomal protein. Notably absent is a significant inflammatory response. Within the lung, LAM nodules are directly adjacent to areas of cystic change. Lining these cystic spaces, the aetiology of which is unclear, are hyperplastic type II cells, in contrast to the hyperplastic type I cells present in emphysema.109 Isolated type II cell hyperplasia may also occur in the absence of LAM nodules. In tuberous sclerosis, type II pneumocytes form clusters termed multifocal micronodular pneumocyte hyperplasia that are unique to TSC and may occur in the absence of LAM in these patients. These lesions are not associated with mutations in TSC2.110
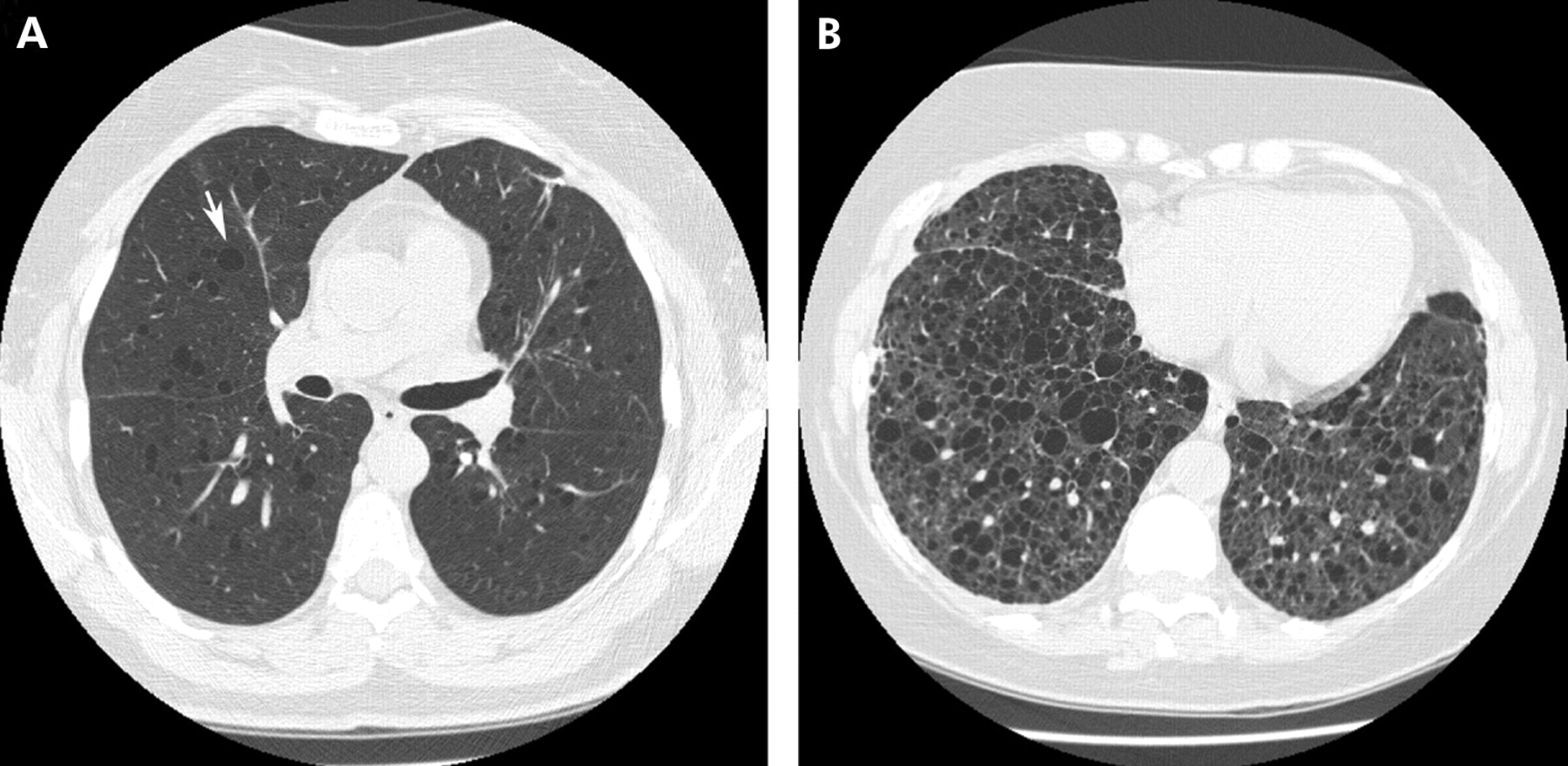
Plain chest radiographic abnormalities in patients with LAM may be mild or absent at presentation, depending on the severity of the disease. The chest radiograph is non-specific, demonstrating normal to hyperinflated lung volumes with bilateral reticular opacities (fig 9). Small nodules may be present but, in contrast to PLCH, they are not as diffuse. Pneumothoraces, which are present in up to 50% of patients at presentation, occur in up to 80% of patients during the course of their disease.111 Pleural effusions, typically chylous, occur in up to 20% of patients. With advanced disease, diffuse cysts and bullae may occur. HRCT imaging in the appropriate clinical setting is diagnostic.112 It shows thin-walled cysts of variable size diffusely distributed throughout all lung fields (fig 9). The cysts may vary in size from 2 to 40 mm. Vessels can be identified at the periphery of the cysts, in contrast to emphysema in which vessels are present within the centre of the lesion.113 The cyst walls are 1–2 mm in thickness and, in the absence of high-resolution imaging, patients may be misdiagnosed as having emphysema. Septal thickening occurs but is atypical and may represent lymphatic dilation due to smooth muscle proliferation and obstruction.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High-resolution CT scans from two patients with lymphangioleiomyomatosis (LAM). Diffuse, discrete, uniform cysts (arrow) are present early in the course of the disease (A). As the disease progresses, cysts may coalesce to form larger cysts (B). In contrast to pulmonary Langerhans’ cell histiocytosis, nodules are not a prominent feature in LAM.
The natural history of LAM is variable and therapeutic options for its treatment are controversial. No medical treatment has been proved to be beneficial. In those with severe progressive disease, lung transplantation remains the cornerstone of treatment. While some patients may respond to hormone manipulation, most appear resistant to this approach.114–116 We do not generally recommend hormonal manipulation in our patients, but do treat reversible airflow limitation with inhaled β agonists and recommend pleurodesis in those with recurrent pneumothoraces. Animal studies suggest that rapamycin, a peptide isolated from Streptomyces hygroscopicus with potent immunosuppressive properties, may have some role in treatment in the future. An early phase trial investigating the efficacy of rapamycin in the treatment of AMLs in tuberous sclerosis and LAM is in progress.117
CONCLUSIONS
While two diffuse (PLCH and LAM) and two focal or multifocal (DIP and LIP) cystic lung diseases are the most commonly considered problems, cystic lung disease may be caused by a variety of congenital and acquired lung diseases. As the prognosis—and therefore evaluation and subsequent management—may fundamentally differ among them, a confident diagnosis is important. Separation of cystic disease from its mimics such as cavities, emphysema and honeycombing is necessary in this regard. The clinical and, more important, the HRCT features of wall thickness, size, overall number and anatomical distribution can provide a high degree of confidence in a particular diagnosis or limit the differential diagnosis to only a few possibilities.
REFERENCES
Footnotes
-
This work is supported by the NIH (NHLBI SCOR HL67671).
-
Competing interests: None.
-
Dr Cosgrove is a Parker B Francis Fellow in Pulmonary Research.