Article Text

Abstract
Rationale: Little knowledge exists on structural changes and plugging in small airways in cystic fibrosis.
Objective: To characterise the extent of plugging and contribution of secreted mucins to the plugs.
Methods: Small airways in patients with cystic fibrosis at transplantation (n = 18) were compared with control non-smokers (n = 10). Tissue sections were stained with Alcian blue (AB)/periodic acid-Schiff (PAS), for mucins MUC5B and MUC5AC, and for neutrophils and its chemoattractant interleukin (IL) 8. Epidermal growth factor receptor (EGFR) and its ligand pro-transforming growth factor α were also identified using immunohistochemical staining. Epithelial and luminal contents were assessed morphometrically.
Results: Plugs occupying >50% of total luminal volume were found in 147 of 231 (63.6%) airways in patients with cystic fibrosis, but only in 1 of 39 (2.6%) airways in controls. In the epithelium of patients with cystic fibrosis, AB/PAS, MUC5B, and MUC5AC-stained volume densities were increased 10-fold (p<0.01), indicating increased mucin production. In airway lumens, staining for mucins was also increased in cystic fibrosis, indicating increased mucin secretion. In the epithelium of patients with cystic fibrosis, neutrophil numbers were markedly increased and were inversely correlated with volume densities of mucous glycoconjugates (r = −0.66, p<0.005). IL8 staining was increased in the epithelium of patients with cystic fibrosis and colocalised with mucins. Staining for EGFR and for pro-transforming growth factor α were increased in the epithelium of patients with cystic fibrosis; positive correlations were found between EGFR-stained volume density and both AB/PAS and IL8-stained volume densities.
Conclusions: Most of the small airways are plugged in cystic fibrosis at the time of transplantation. Mucins contribute to airway plugging. Recruited neutrophils may be involved in mucin secretion in the plugs. Increased expression of EGFR and its ligand suggests roles in mucin synthesis and neutrophil recruitment.
- AB/PAS, Alcian blue/periodic acid-Schiff
- EGFR, epidermal growth factor receptor
- TGF, transforming growth factor
Statistics from Altmetric.com
- AB/PAS, Alcian blue/periodic acid-Schiff
- EGFR, epidermal growth factor receptor
- TGF, transforming growth factor
Cystic fibrosis is a genetic disorder caused by mutations of a single gene that encodes for the cystic fibrosis transmembrane regulator protein.1 It remains one of the most common life-shortening genetic disorders.1 Cystic fibrosis-related lung disease is characterised by plugging of airways associated with persistent bacterial infection and massive neutrophil infiltration.1 Plugging in small airways contributes to the morbidity and mortality in cystic fibrosis, leading to respiratory failure and the need for lung transplantation.2
Progress in the understanding of the pathobiology of small airway plugging in cystic fibrosis has been impeded by several factors. Firstly, no adequate animal model of cystic fibrosis airway disease is available.3 Secondly, because of their peripheral location, examination of these airways in live patients has been difficult and rare. We took advantage of the opportunity to examine small airways in the lungs of patients with cystic fibrosis to find and quantify morphological changes at the time of transplantation, acknowledging the fact that the effects in these lungs are due to a life-time of insults. Nevertheless, we recognised that the disease is progressive and reasoned that even at the time of transplantation, important information could be obtained regarding the pathobiology of the disease. The purpose of this study is to obtain structural information regarding potential pathways for airway plugging, mucin production and neutrophil recruitment that we hope will help in designing future mechanistic studies in patients with cystic fibrosis.
Mucins are complex glycoproteins characterised by a large molecular size and high carbohydrate contents. The gel-forming mucins MUC5B and MUC5AC are cysteine rich, secreted mucins containing tandem repeats in the protein backbone. These two mucins have been convincingly found in human airway tissue and in human airway secretions using immunohistochemical staining. On hydration in the airway lumen, these mucins form a viscoelastic gel that normally helps in the removal of foreign material and bacteria.
In this study, we characterised plugging in the small airways in patients with cystic fibrosis. Since a recent study has challenged the contribution of mucins to hypersecretion in the airways of patients with cystic fibrosis,4 we examined the presence of mucous glycoconjugates and of the two major gel-forming mucins, MUC5B and MUC5AC, in the epithelium and in the adjacent plugs. Previous studies have shown that neutrophil products, especially neutrophil elastase, cause rapid goblet cell degranulation.5,6,7 Experimentally, activated neutrophils in contact with epithelial goblet cells cause mucin secretion into the lumen.6 Thus, we studied the localisation of neutrophils and their major epithelial chemoattractant interleukin (IL) 8 in relation to mucins in the airway epithelium. As studies have shown that activation of epidermal growth factor receptors (EGFR) results in mucin synthesis8 and production of IL8 in experimental models,9 we also studied the presence and localisation of EGFR and its ligand pro-transforming growth factor (TGF)-α. In each case, the findings in airways of patients with cystic fibrosis were compared with the findings in control non-smokers.
METHODS
Subjects and tissues
We studied lung specimens obtained at transplantation from 18 non-smoking adults with cystic fibrosis and from 10 non-smoking controls undergoing lung resection for peripheral lung cancer. None of the patients with cystic fibrosis required invasive mechanical ventilation before the lung transplantation procedure. Table 1 shows the clinical characteristics of the patients. No significant difference for any outcome was found among patients with various cystic fibrosis transmembrane regulator genotypes (not shown). To reduce the potential sampling bias related to irregular distribution of epithelial changes in patients with cystic fibrosis, tissues were obtained from two to four blocks (depending on tissue availability), whose locations were chosen randomly. In controls, only one block of tissue removed in a macroscopically normal area at a distance from the cancer was available for study. Additional information regarding tissue selection is provided in a supplementary file available at http://thorax.bmjjournals.com/supplemental. The study conformed to the Declaration of Helsinki and to the rules of the Committee on Human Research of Hôpital Cochin, Paris, France.
Clinical characteristics of patients
Histochemical and immunohistochemical staining
Paraffin tissue sections 5 μm in size from blocks that contained small airways (defined as non-cartilaginous airways surrounded by lung parenchyma) when examined with haematoxylin-eosin were stained with Alcian blue (AB)/periodic acid-Schiff (PAS) to identify mucous glycoconjugates.
We performed immunohistochemical staining for mucins MUC5B and MUC5AC, human neutrophil elastase (to identify neutrophils), IL8, EGFR and pro-TGF-α, using previously described methods.10 The following antibodies were used: a polyclonal antibody to MUC5B raised against a recombinant protein corresponding to amino acids 1201–1500 of MUC5B (clone H300; dilution 1:200; Santa Cruz Biotechnology, Santa Cruz, California, USA) was used to identify and quantify MUC5B protein in airways of patients with cystic fibrosis; a second polyclonal antibody to MUC5B (LUM5B-2, which recognises peptides present in the cystein-rich domains of the super repeats in the central exon of MUC5B protein,11 a kind gift of Pr I Carlstedt, Sweden) was used to confirm the surprising finding that MUC5B was present in the small airway epithelium using clone H300; a monoclonal antibody to MUC5AC (clone 45M1; dilution, 1:500; Neomarkers, Fremont, California, USA); a monoclonal antibody to human neutrophil elastase (NP57; dilution, 1:100; Dako, Carpinteria, California, USA); a monoclonal antibody to EGFR (dilution, 1:600; Calbiochem, La Jolla, California, USA); a monoclonal antibody to pro-TGF-α (antibody2; dilution, 1:50; Calbiochem); and a polyclonal antibody to IL8 (dilution, 1:20; Biosource, Camarillo, California, USA). Omission of the primary antibody or incubation with irrelevant immunoglobulins of the same isoptype were used as negative controls. A full description of the immunohistochemical protocols is provided in a supplementary file available at http://thorax.bmjjournals.com/supplemental.
Quantification of airway dimensions and luminal contents
A total of 39 airways in controls (mean, 3.9 airways/subject) and 231 airways in patients with cystic fibrosis (mean, 12.8 airways/patient) were analysed. The median (min–max) number of airways per patient examined was 4 (1–7) in controls and 12 (7–30) in patients with cystic fibrosis. We used the NIH image software (available on line at http://rsb.info.nih.gov/ij/) to determine airway dimensions by measuring internal perimeters, defined as the luminal border of the airway.12 The mean (SD) internal perimeter was 1.8 (1.5) mm in controls and was increased in airways of patients with cystic fibrosis (6.3 (8.9) mm, p<0.05), a finding consistent with the occurrence of bronchiectasis in cystic fibrosis. Total luminal volumes were measured by point counting using a grid. Points within the grid falling on airway lumens were counted; each point corresponds to a known volume allowing for determination of total luminal volume.13 Similarly, points within the grid falling on luminal contents stained with haematoxylin-eosin or with AB/PAS, MUC5B and MUC5AC were counted. Results are expressed as ratios of luminal volumes stained for haematoxylin-eosin, AB/PAS, MUC5B or MUC5AC to total luminal volumes (%). Airway lumens that contained ⩾50% of their lumens stained with haematoxylin-eosin were defined as plugged airways. Additional information is provided in an online data supplement.
Quantification of AB/PAS, MUC5B, MUC5AC, IL8, EGFR and pro-TGF-α staining in epithelium
Measurements of volumes occupied by staining for AB/PAS, MUC5B, MUC5AC, IL8, EGFR and pro-TGF-α in airway epithelium were performed by point counting using a grid.13 Points within the grid falling on epithelial cells staining positively were counted. As we found that the total epithelial volume was increased in patients with cystic fibrosis compared with control (see table A, in the online data supplementary file at http://thorax.bmjjournals.com/supplemental), volumes occupied by positively-stained cells were normalised per surface unit of basal membrane, which did not change between the two groups. Thus results are expressed as stained volumes/surface of basal membrane (volume densities), as described previously.10,13 However, the conclusions were similar whether the total volume of epithelium or the surface of basal membrane was used as the reference compartment (see table A in the online supplementary file at http://thorax.bmjjournals.com/supplemental). In each patient with cystic fibrosis, these studies were conducted in 30 randomly selected high power fields (magnification×400) of epithelium from two to four sections obtained in randomly selected blocks (see above). In controls, quantification was performed in a single block; we analysed a median (range) number of 18 (4–30) high power fields. The median (min–max) length of epithelium analysed for each staining in patients with cystic fibrosis was 5.19 (2.77–5.42) mm and was 3.68 (1.25–5.89) mm in controls.
Relationship between neutrophils and mucous glycoconjugates in epithelium
The relationship between neutrophil numbers and mucous glycoconjugates in airway epithelium was evaluated in sections stained for human neutrophil elastase and counterstained with AB/PAS. Human neutrophil elastase-stained cells were counted in epithelium in 30 randomly selected high power fields (magnification×400). Results were expressed as the number of cells/mm of basal membrane. Volume densities occupied by mucous glycoconjugates were measured by point counting in the same fields (see above).
Statistical analysis
Data are expressed as mean (standard error of mean (SEM)), unless otherwise specified. Data obtained from morphometric measurements were analysed using the non-parametric Kruskal–Wallis test followed by Dunn’s test for multiple comparisons, the Mann Whitney U test and the Spearman’s rank correlation. Differences were considered significant for p<0.05. Analyses were performed using Graphpad Prism 4.
RESULTS
Small airway plugging
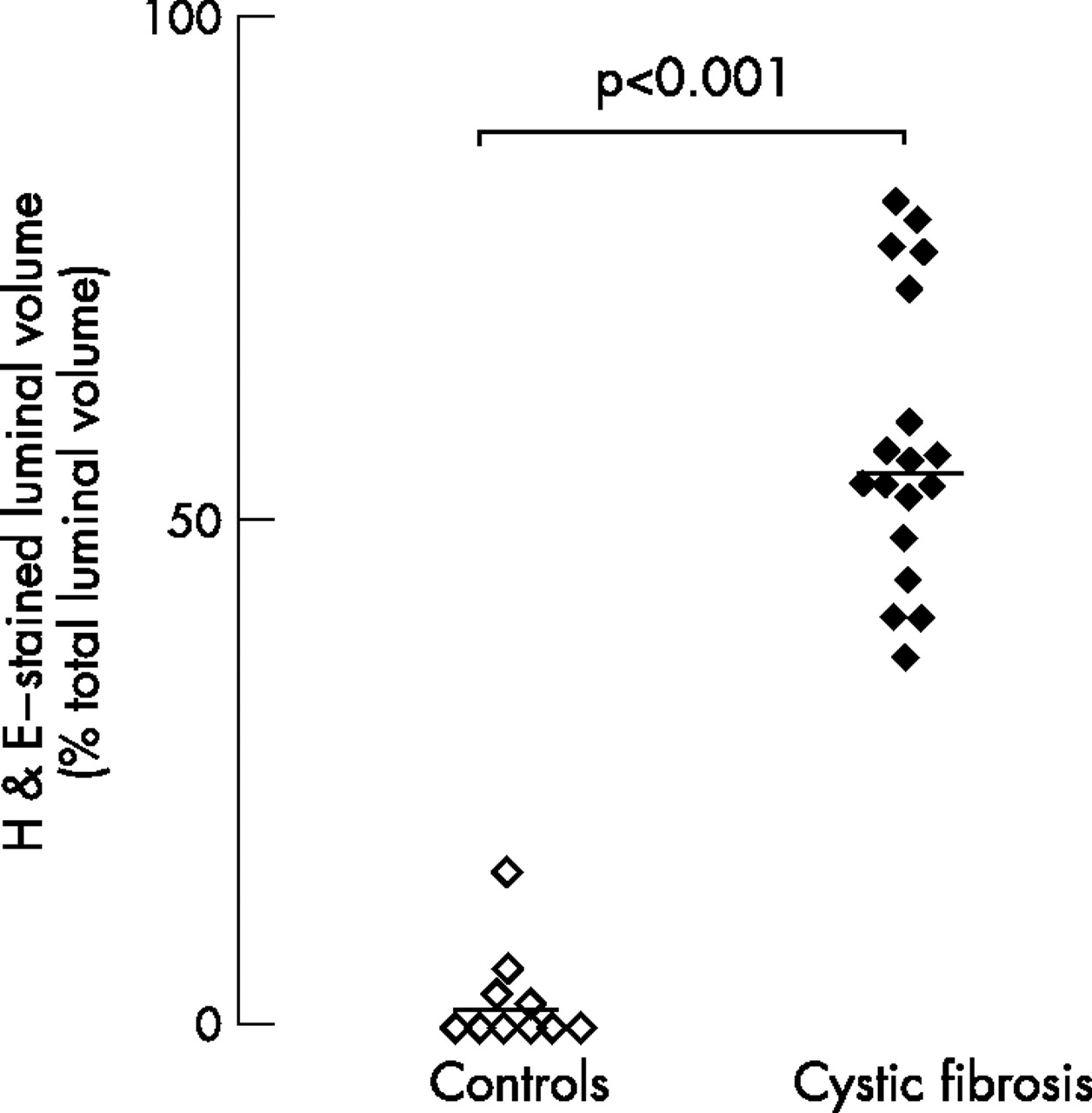
In control subjects (n = 10), airway lumens contained little or no staining with haematoxylin-eosin (empty lumens). However, in all patients with cystic fibrosis (n = 18), there was extensive staining with haematoxylin-eosin in airway lumens. Thus, volume ratios of luminal haematoxylin-eosin staining to total luminal volumes were increased in airways of patients with cystic fibrosis compared with controls (p<0.001; fig 1). Plugs occupying >50% of total luminal volume were found in 147 of 231 (63.6%) airways in patients with cystic fibrosis, but only in 1 of 39 (2.6%) airways in controls.

Luminal contents in airways in controls and in patients with cystic fibrosis. Sections from control subjects (open symbols), and patients with cystic fibrosis (solid symbols) were stained with haematoxylin-eosin (H&E). Volumes of contents stained with H&E in airway lumens were measured and expressed as ratios of stained luminal volume to total luminal volume (%). Horizontal bars represent median values.
Mucous glycoconjugates and mucins MUC5B and MUC5AC in epithelium and lumen
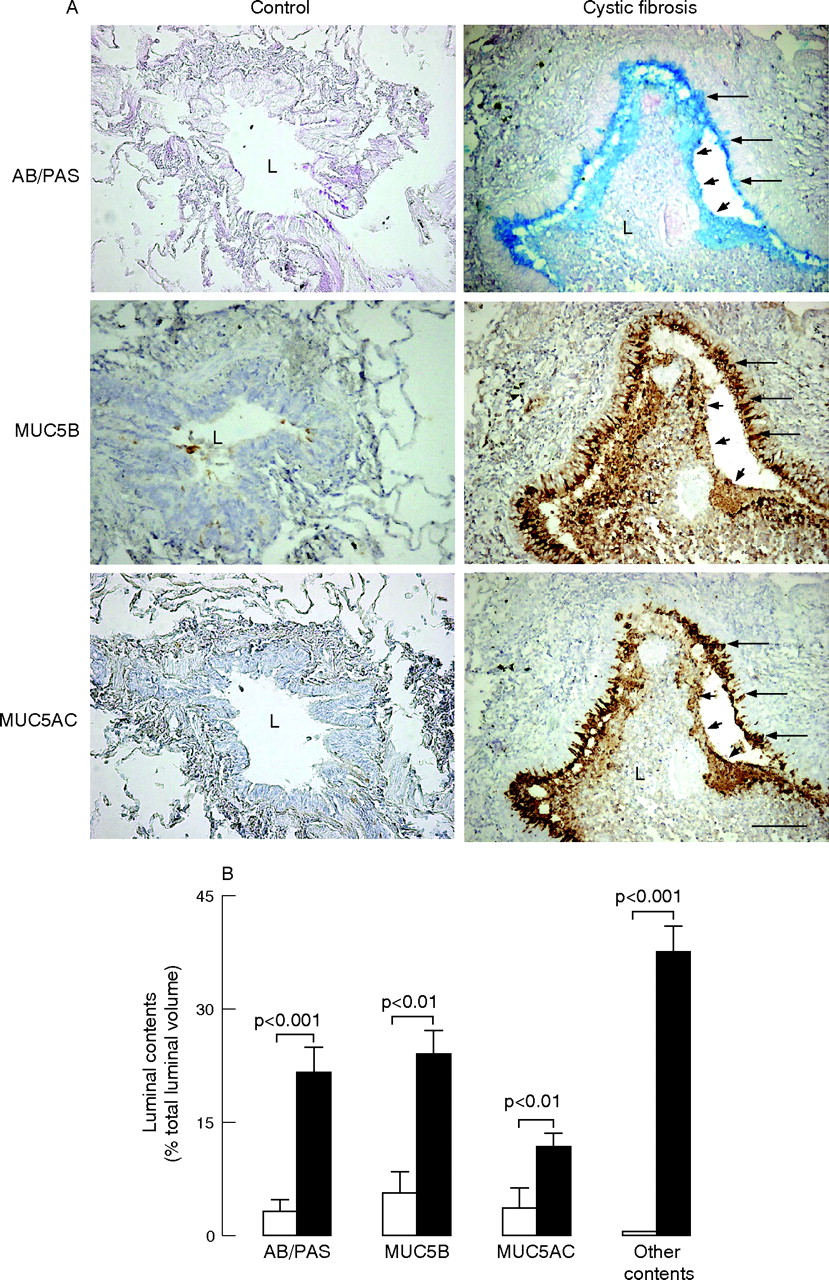
In epithelium, both AB/PAS staining for mucous glycoconjugates and immunostaining for mucins MUC5B and MUC5AC were consistently sparse in control subjects but were markedly increased in patients with cystic fibrosis (each comparison, p<0.01; fig 2). The volume densities occupied by AB/PAS, MUC5B and MUC5AC staining in the epithelium were not significantly different from one another in each group of subjects (each comparison, p>0.10). In the epithelium, MUC5AC and MUC5B colocalised in most goblet cells.

Quantification of mucous glycoconjugates and mucins MUC5B and MUC5AC in the epithelium of controls (open symbols) and patients with cystic fibrosis (solid symbols). Sections were stained with Alcian blue (AB)/periodic acid-Schiff (PAS) for identification of mucous glycoconjugates or with antibodies to MUC5B or MUC5AC mucin. Quantification was performed by morphometric point counting (see Methods). Results are reported as stained volume/mm2 of basal membrane (BM). Horizontal bars represent median values.
In airway lumens of patients with cystic fibrosis, AB/PAS staining and mucin MUC5B and MUC5AC staining were present widely and were mostly concentrated in the periphery of the plugs adjacent to the epithelium (fig 3A). Other contents including inflammatory cells, especially neutrophils, but also macrophages, DNA staining and bacterial macrocolonies (not shown), were seen throughout lumens forming muco-inflammatory plugs. In airway lumens, volumes occupied by mucous glycoconjugates, MUC5B and MUC5AC were increased in patients with cystic fibrosis compared with controls, indicating increased mucin secretion (p<0.01, fig 3B). Similarly, volumes occupied by other contents were increased in patients with cystic fibrosis compared with controls. In lumens of patients with cystic fibrosis, volumes occupied by glycoconjugates were smaller than volumes occupied by other contents (p<0.01), and volumes occupied by MUC5AC were smaller than volumes occupied by MUC5B or by AB/PAS (p<0.005, fig 3B).

Mucous glycoconjugates and mucins in airway lumens of controls and patients with cystic fibrosis. (A) Representative photomicrographs. Sections containing small airways in a typical control subject (left panels) and in a patient with cystic fibrosis (right panels) were stained with Alcian blue/periodic acid-Schiff (AB/PAS (for mucous glycoconjugates, blue staining, upper panels)) or with antibodies to mucin MUC5B (brown staining; middle panels) and MUC5AC (brown staining, lower panels). In the control subject, the airway lumen (L) is empty; no staining for AB/PAS, MUC5B or MUC5AC is observed in the lumen or in the epithelium. In the patient with cystic fibrosis, the airway lumen is obstructed by a plug containing mucous glycoconjugates (AB/PAS), MUC5B and MUC5AC only at the periphery of the plug (arrowheads), adjacent to the epithelium. Staining for mucins is absent in the centre of the plug. Goblet cells in the epithelium are stained positively with AB/PAS and with MUC5AC and MUC5B antibodies (arrows). Original magnification×100, bar 100 μm. (B) Quantification of mucous glycoconjugates and mucins MUC5B and MUC5AC in airway lumens. Sections were stained with AB/PAS or with antibodies to MUC5B and MUC5AC. In the airways of controls (n = 10, open bars) and in airways of patients with cystic fibrosis (n = 18, solid bars), volumes occupied by AB/PAS, MUC5B and MUC5AC staining in airway lumens were measured and expressed as ratios of total luminal volume (%; see Methods). Similarly, volumes of luminal contents that were not stained with AB/PAS (other contents) in airway lumens were determined. Results are shown as mean (SEM).
Neutrophil recruitment and relationship to mucous glycoconjugates
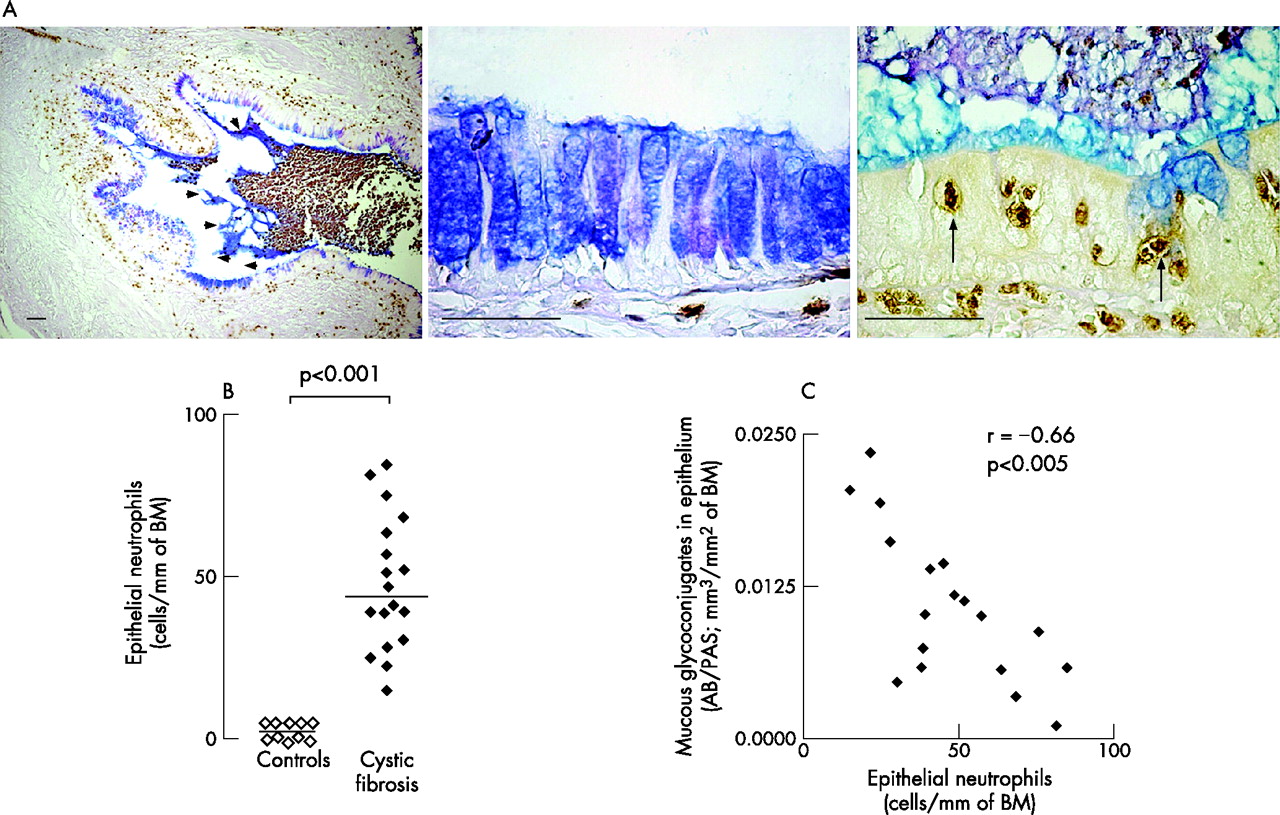
Neutrophil numbers were low in the airway epithelium of controls. In airways of patients with cystic fibrosis, neutrophils were increased markedly in lumens (fig 4A, left panel) and in the epithelium (p<0.001; fig 4B). We analysed the relationship between neutrophils and mucous glycoconjugates in sections stained for elastase and AB/PAS. When large numbers of neutrophils were found in an area of epithelium, epithelial mucous glycoconjugates were sparse and were found in the airway lumen streaming from epithelial cells (fig 4A, right panel). Conversely, when large numbers of goblet cells were present in an area of epithelium, few or no neutrophils were observed in the epithelium (fig 4A, middle panel). Thus, neutrophil numbers in the epithelium were inversely correlated with volume densities occupied by mucous glycoconjugates (r = −0.66; p<0.005; n = 18, fig 4C).

Relationship between mucins and neutrophils. (A) Localisation of neutrophils in airways of patients with cystic fibrosis. Relationship to mucous glycoconjugates. Sections were stained with a monoclonal antibody to human neutrophil elastase (brown staining) and counterstained with Alcian blue/periodic acid-Schiff (AB/PAS; blue staining) for mucous glycoconjugates. Large numbers of neutrophils are seen in the airway lumen and AB/PAS in the lumen is localised to the periphery of the plug (left panel, arrowheads). In the epithelium where large amounts of mucous glycoconjugates are present, no neutrophils are found in the epithelium (middle panel). In areas where large numbers of neutrophils are found in the epithelium (right panel, arrows), only small amounts of mucous glycoconjugates are present in the epithelium, but large amounts of mucous glycoconjugates are present in the lumen adjacent to the epithelial cells, suggesting goblet cell degranulation. Original magnifications are from ×50 to ×400. Bars 50 μm. (B) Quantification of epithelial neutrophils in controls (open symbols) and in patients with cystic fibrosis (solid symbols). Neutrophils (identified as elastase-stained cells) were counted in epithelium (see Methods) and reported as the number of neutrophils/mm of basal membrane (BM). Horizontal bars represent median values. (C) Relationship between neutrophil numbers and mucous glycoconjugates in the epithelium of patients with cystic fibrosis. Sections were double-stained with a monoclonal antibody to human neutrophil elastase and with AB/PAS (for mucous glycoconjugates). Neutrophils were counted in epithelium and AB/PAS-stained volumes were measured as described in the Methods. Each symbol corresponds to data obtained in one patient with cystic fibrosis.
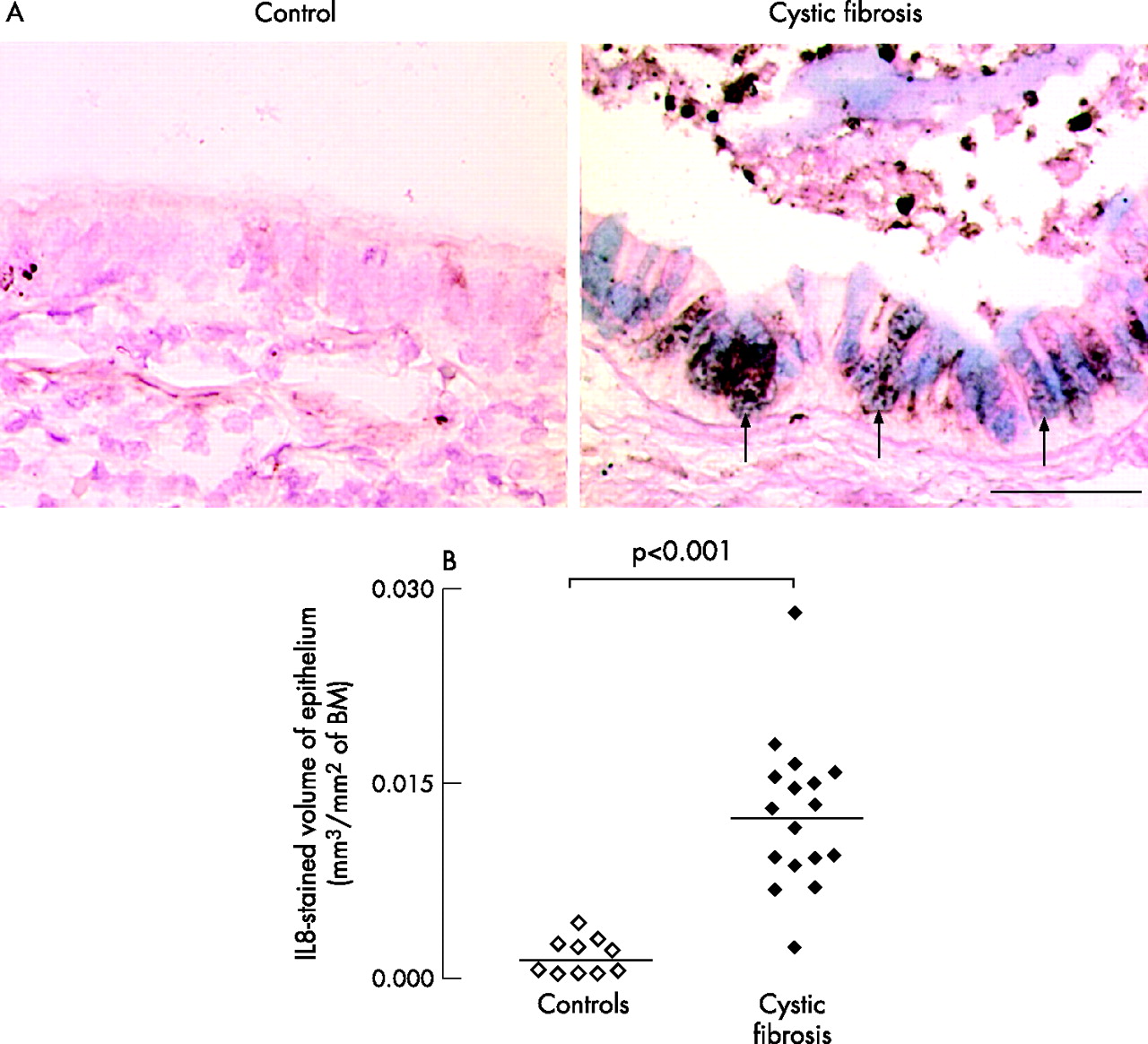
Staining for the neutrophil chemoattractant IL8 was prominent in the epithelium of patients with cystic fibrosis (fig 5A), and in recruited neutrophils, especially in the airway lumen, but IL8 staining was sparse in the epithelium of controls. In the epithelium, IL8 colocalised with AB/PAS in goblet cells was not expressed in basal or ciliated cells. Volume densities occupied by IL8-stained cells in the epithelium were increased in patients with cystic fibrosis compared with controls (p<0.001, fig 5B).
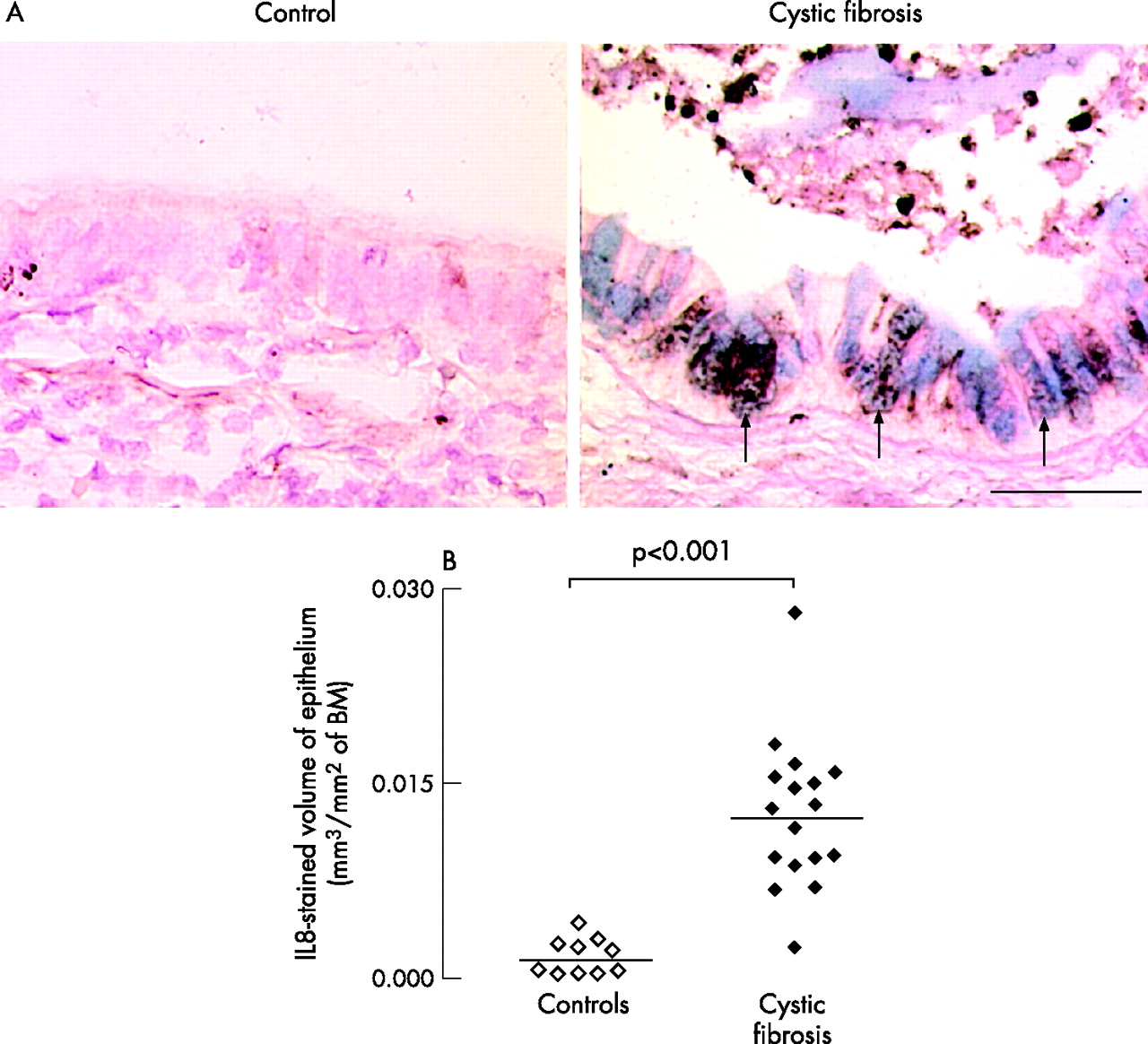
Presence and localisation of the neutrophil chemoattractant interleukin (IL) 8 in the epithelium of controls and patients with cystic fibrosis. (A) Representative photomicrographs. Sections were stained with a polyclonal antibody to IL8 (brown staining) and counterstained with Acid blue/periodic acid-Schiff (AB/PAS) for mucous glycoconjugates (blue staining). In the epithelium of the control subject, no staining for IL8 or AB/PAS is found. In the epithelium of patients with cystic fibrosis, both IL8 and AB/PAS staining are prominent and often colocalise (arrows). Neutrophils in the airway lumen also stain positively for IL8. Original magnification 400×. Bar 50 μm. (B) Quantification of epithelial IL8 in controls (open symbols) and in patients with cystic fibrosis (solid symbols). Sections were stained with an antibody to IL8 and data were obtained using morphometric point counting (see Methods). Results are reported as stained volume/surface of basal membrane (BM). Horizontal bars represent median values.
EGFR and pro-TGF-α in epithelium
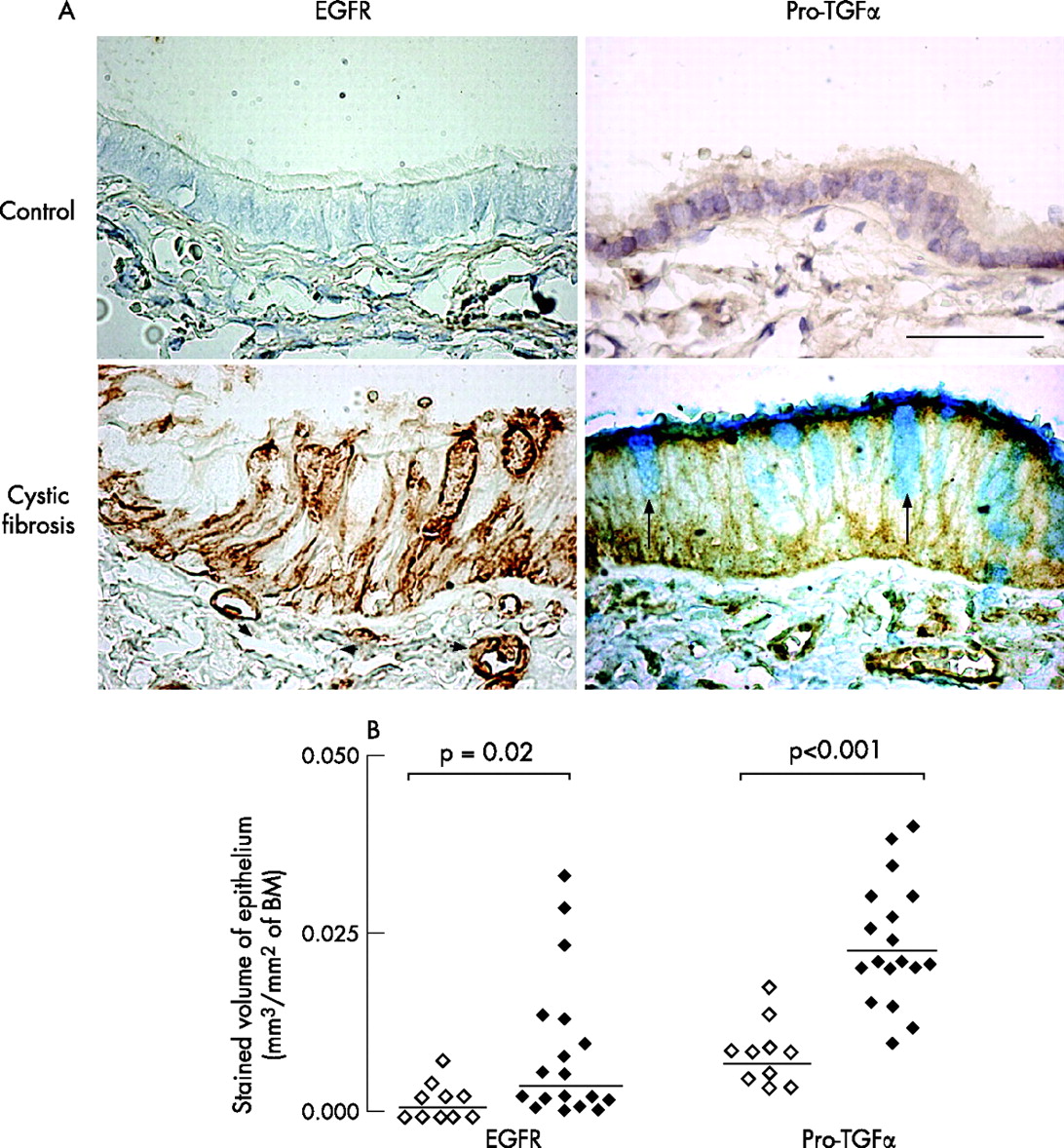
In the epithelium of controls (n = 10), little or no staining for EGFR protein was found (fig 6A). In patients with cystic fibrosis, EGFR was strongly expressed in epithelium and in submucosal blood vessels in some subjects (fig 6A), whereas expression of EGFR was low in other subjects. Staining for EGFR was present in basal cells and in some elongated non-ciliated cells (presumably Clara cells), but was absent in ciliated cells. Stained volume densities of EGFR in epithelium were significantly increased in patients with cystic fibrosis (p = 0.02; fig 6B). In patients with cystic fibrosis, volume densities of epithelium stained for EGFR were positively correlated with both AB/PAS- and IL8-stained volume densities (r = 0.54, p = 0.02 and r = 0.51, p = 0.04, respectively; n = 18).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Epidermal growth factor receptor (EGFR) and pro-transforming growth factor (TGF)α staining in epithelium. (A) Representative photomicrographs of EGFR and pro-TGFα immunostaining in a control (upper panels) and in a patient with cystic fibrosis (lower panels). Sections were stained with monoclonal antibodies to EGFR (left columns) and pro-TGFα (right columns). In the control subject, no EGFR is found in the epithelium or in blood vessels. In the patient with cystic fibrosis, brown staining for EGFR is seen in epithelial basal cells and columnar non-ciliated cells; intense staining is also observed in subepithelial blood vessels (arrowheads). Staining for pro-TGF-α is only faint in the control subjects, whereas strong brown staining is observed in both epithelial cells and in subepithelial blood vessels. Counterstaining with AB/PAS (blue staining) shows that goblet cells (arrows) do not contain pro-TGFα. Original magnification 400. Bar = 50 μm. (B) Quantification of EGFR and pro-TGFα immunoreactivity in the epithelium of controls (open symbols) and patients with cystic fibrosis (solid symbols). Sections were stained with monoclonal antibodies to EGFR and pro-TGFα, and data were obtained using morphometric point counting (see Methods). Results are reported as positively-stained volume/mm2 of basal membrane (BM). Horizontal bars represent median values.
Immunostaining for pro-TGFα, a major EGFR ligand in airways, was faint in the epithelium of controls. The expression of pro-TGFα was increased markedly in the epithelium in airways of patients with cystic fibrosis compared with controls (p<0.001, n = 18; fig 6A, 6B). Staining for pro-TGFα was present in epithelial ciliated cells, but absent in goblet cells. In airways of patients with cystic fibrosis, large numbers of inflammatory cells, which seemed to be macrophages (not shown), also expressed pro-TGF-α. In the epithelium, no correlation was found between pro-TGF-α and AB/PAS staining (not shown).
DISCUSSION
The development of small airway obstruction by plugging is believed to have a major role in the pathophysiology of cystic fibrosis,1 but to our knowledge the extent and characteristics of plugging have never been quantified. In this study, we took advantage of the opportunity to analyse structural features of small airways in the lungs of patients with cystic fibrosis removed at the time of transplantation. Small airways of control subjects showed only rare plugging, whereas most small airways of patients with cystic fibrosis contained extensive plugging.
In this study, our group of patients with cystic fibrosis was composed of subjects with advanced cystic fibrosis undergoing lung tranplantation with relatively homogenous clinical characteristics (low forced expiratory volume in the first second, colonisation with Pseudomonas aeruginosa). Thus, we were unable to correlate the extent of small airway plugging to lung function. However, our data show that in subjects with advanced lung disease (low forced expiratory volume in the first second and forced vital capacity), most small airways are plugged, suggesting that small airway plugging contributes to disease progression (loss in lung function) in cystic fibrosis.
Although subjects in our control group were older than patients in the cystic fibrosis group, we do not believe that this difference is likely to alter the conclusions. Further, because the tissues were fixed using endobronchial inflation,14 a standard procedure for lung fixation that may result in the loss of luminal contents, our morphological evaluation of plugging is likely to underestimate the extent of plugging.
We sought information from the lung structure of patients with cystic fibrosis on potential mechanisms involved in progressive plugging of the airways by analysing the components of the plugs and the adjacent epithelium. Here we found a striking increase in mucous glycoconjugates (recognised by AB/PAS staining), and in MUC5B and MUC5AC mucin staining in the small airway epithelium of patients with cystic fibrosis, often associated with plugging of the lumens. Earlier authors suggested that mucins contribute to hypersecretion in cystic fibrosis,15–17 but no study has assessed quantitatively the staining for mucin proteins in airways of patients with cystic fibrosis. Recently, Henke et al4 reported that concentrations of MUC5AC and MUC5B are decreased in the airway secretions of patients with cystic fibrosis. The discrepancy between these findings and our study may result from several explanations. Firstly, Henke et al compared two entirely different secretions (sputum of patients with cystic fibrosis and materials harvested from endotracheal tubes of normals); thus, the two samples are difficult to compare. Secondly, sampling of sputum does not discriminate the source of secretions, especially from peripheral airways. Thus, the sampling by Henke et al provides no information concerning the contents of plugs in the periphery of the lungs. Our study clearly shows profound goblet cell hyperplasia and mucins in the luminal plugs in small airways of patients with cystic fibrosis. However, it should be mentioned that there was some variability in the epithelial staining for mucins among patients with cystic fibrosis (fig 2) and among different airways in the same patient with cystic fibrosis (not shown). Further, although mucins clearly have a role in the formation of the plugs in cystic fibrosis, other components (eg, recruited neutrophils, DNA largely originating from dead neutrophils and bacterial macrocolonies) also contribute to the formation of the plugs.
The presence of MUC5AC in epithelium is not surprising because it has been shown to be a predominant mucin in airway epithelium.13,18 However, the finding of MUC5B in the epithelium of patients with cystic fibrosis deserves comment: this mucin has been considered to be predominantly expressed in glands,19 but it was also reported recently in the epithelium in diffuse panbronchiolitis.20 The finding of MUC5B protein in the epithelium of patients with cystic fibrosis is also consistent with the report of MUC5B mRNA in the bronchiolar epithelium of patients with emphysema and pulmonary fibrosis.21 By performing double immunostaining with MUC5AC and MUC5B antibodies, we found that most surface epithelial goblet cells stained positively (not shown). The biophysical differences of different mucins and their implications are not yet understood and deserve further study.
In plugged airway lumens, staining with AB/PAS and with MUC5B and MUC5AC antibodies showed mucins streaming from epithelial cells into luminal plugs. Morphometric analysis showed that AB/PAS staining occupied approximately 20% of the lumen volume, indicating that mucins contribute significantly to the plugging process. Serine proteases have been reported to degrade mucins.22 Thus, Voynow et al23 showed that MUC5AC is degraded in vitro in the presence of purified neutrophil elastase. Davies et al24, measuring MUC5AC and MUC5B concentrations in the sputum of patients with cystic fibrosis, found increased MUC5AC compared with MUC5B in the sol phase of sputum, suggesting increased proteolytic degradation of MUC5AC. We found that MUC5B was more prominent in the lumen of patients with cystic fibrosis than MUC5AC, whereas staining for these mucins was comparable in the epithelium. The predominance of MUC5B in the airway lumen may reflect the greater susceptibility of MUC5AC to proteolytic degradation in the lumen when compared with MUC5B.24
Neutrophil infiltration in airways is known to play important roles in cystic fibrosis-airway disease.25 We found prominent neutrophil recruitment in the epithelium of patients with cystic fibrosis, confirming a previous study.26 In this study, in the epithelium of patients with cystic fibrosis, neutrophils were localised to the areas where mucins were sparse in the epithelium and were seen streaming from epithelial (goblet) cells into the lumen. Neutrophil elastase is known to cause rapid and potent degranulation of goblet cell mucins.5,6 The finding that neutrophils were absent from the areas of epithelium containing intact goblet cells and the presence of mucins streaming into the lumens in areas of epithelium containing neutrophils suggests that neutrophils may be a potent source of goblet cell degranulation in cystic fibrosis. Neutrophil products have also been implicated in mucin production by airway epithelial cells.23,27 However, mucin production is a slow process requiring up to 24 h to complete,28 long after the neutrophils are predicted to have moved out of the epithelium. By contrast, mucin degranulation is a rapid process occurring within seconds to minutes. Thus, the finding that mucins were absent when neutrophils were found in epithelium does not preclude a role for neutrophils in mucin synthesis. Furthermore, as suggested above, neutrophils may also have a role in the degradation of mucins in the airway lumen. Thus, in cystic fibrosis, neutrophils are likely to be involved in multiple processes related to mucin production, secretion and degradation.
A wide variety of stimuli present in the airways of patients with cystic fibrosis (eg, Pseudomonas aeruginosa products) are known to cause mucin overproduction by EGFR activation.29Pseudomonas aeruginosa products also cause epithelial expression and secretion of the neutrophil chemoattractant IL8.30 Richter et al9 reported that the activation of EGFR by its ligands and by cigarette smoke induces IL8 in airway cells. Because we found that IL8 and AB/PAS were colocalised in the airway epithelium of patients with cystic fibrosis, we hypothesise that both mucin and IL8 synthesis in cystic fibrosis could be caused by EGFR activation. Immunostaining for EGFR was consistently sparse or absent in controls, but was increased though variable in patients with cystic fibrosis. Although the reasons for EGFR variability in the epithelium of patients with cystic fibrosis are unknown, the positive correlations between EGFR staining and both IL8 and AB/PAS staining suggest possible causal relationships. Further, we show that a prominent EGFR ligand, pro-TGF-α, is markedly increased in the epithelium of patients with cystic fibrosis, confirming a previous report.31 Thus, epithelial cells in patients with cystic fibrosis contain the molecules required for mucin and IL8 production.
In summary, small airways in advanced cystic fibrosis are characterised by extensive airway obstruction with muco-inflammatory plugs. Mucins clearly contribute to these plugs. The preferential localisation of mucins adjacent to the epithelium and the inverse relationship between the presence of neutrophils and mucins in epithelium suggest that neutrophil-dependent mucin secretion contributes to plugging. We suggest that the paucity of mucins in the centre of the plugs could be due to the cleavage and degradation of mucins by enzymes (eg, from neutrophils), but proof of this hypothesis will require further studies. Furthermore, the colocalisation of mucin and IL8 suggests that a common mechanism could be responsible for their production in airways of patients with cystic fibrosis. These findings provide important structural information that suggest possible mechanisms involved in the pathogenesis of cystic fibrosis and possible therapeutic strategies.
Acknowledgments
We thank Iris F Ueki for expert technical help and thoughtful discussions, and Professor I Carlstedt (Sweden) who kindly provided the LUM5-B2 antibody.
REFERENCES
Footnotes
-
↵* These authors contributed equally to this work.
-
Published Online First 23 August 2006
-
This work was funded by grants from “Chancellerie des Universités de Paris (Leg Poix)” and “Association Vaincre la Mucoviscidose”. DM received educational grants from Association Cardif and Fonds d’Etude et de Recherche du Corps Médical des Hôpitaux de Paris.
-
Competing interests: None.
-
An abstract of this work was presented at the 2005 ATS meeting, May, San Diego, California, USA.
Linked Articles
- Airwaves