Article Text

Abstract
Backgrounds: Exaggerated bronchial constriction is the most significant and life threatening response of patients with asthma to inhaled stimuli. However, few studies have investigated the contractility of airway smooth muscle (ASM) from these patients. The purpose of this study was to establish a method to measure contraction of ASM cells by embedding them into a collagen gel, and to compare the contraction between subjects with and without asthma.
Methods: Gel contraction to histamine was examined in floating gels containing cultured ASM cells from subjects with and without asthma following overnight incubation while unattached (method 1) or attached (method 2) to casting plates. Smooth muscle myosin light chain kinase protein levels were also examined.
Results: Collagen gels containing ASM cells reduced in size when stimulated with histamine in a concentration-dependent manner and reached a maximum at a mean (SE) of 15.7 (1.2) min. This gel contraction was decreased by inhibitors for phospholipase C (U73122), myosin light chain kinase (ML-7) and Rho kinase (Y27632). When comparing the two patient groups, the maximal decreased area of gels containing ASM cells from patients with asthma was 19 (2)% (n = 8) using method 1 and 22 (3)% (n = 6) using method 2, both of which were greater than that of cells from patients without asthma: 13 (2)% (n = 9, p = 0.05) and 10 (4)% (n = 5, p = 0.024), respectively. Smooth muscle myosin light chain kinase levels were not different between the two groups.
Conclusion: The increased contraction of asthmatic ASM cells may be responsible for exaggerated bronchial constriction in asthma.
- ASM, airway smooth muscle
- MLC, myosin light chain
- MLCK, myosin light chain kinase
- PLC, phospholipase C
- ROCK, Rho-associated coiled coil forming kinase
- smMLCK, smooth muscle MLCK
Statistics from Altmetric.com
- ASM, airway smooth muscle
- MLC, myosin light chain
- MLCK, myosin light chain kinase
- PLC, phospholipase C
- ROCK, Rho-associated coiled coil forming kinase
- smMLCK, smooth muscle MLCK
Excessive airway narrowing to specific or non-specific stimuli is a significant and life threatening feature of asthma. Morphological measurements made on histological preparations of airways from patients with asthma show that the volume of airway smooth muscle (ASM) is increased compared with airways of subjects without asthma.1–4 One theory suggests that the increase in airway wall muscle mass should result in an increased contractile force, which consequently allows for greater narrowing of the airway.5 Despite the fact that several studies clearly show that the ASM mass is increased in asthma, most of the earlier in vitro studies found no difference in the contractile force of bronchi from patients with asthma compared with bronchi from non-asthmatic subjects.6,7 However, Bramley et al observed greater maximal shortening and greater generation of contractile force and stress by ASM strips prepared from asthmatic airways (n = 3) than from non-asthmatic airways (n = 11).8,9 This inconsistency may be attributable to factors such as small sample size and/or a lack of normalisation of force to stress (force divided by smooth muscle cross-sectional area).
It is possible that asthmatic airways do not necessarily generate greater contractile force but still narrow to a greater extent. For example, passively sensitised bronchial tissues from dogs10 and humans11 exhibit greater maximal shortening and maximal shortening velocity without any increase in the maximal contractile force. The reason for this is believed to be an increase in myosin light chain kinase (MLCK) levels which, as a consequence of myosin phosphorylation, controls the rate of cross bridge formation between myosin and actin. Similarly, the maximal shortening and maximal shortening velocity in trypsin-dissociated single ASM cells obtained from endobronchial biopsy specimens from subjects with and without asthma was greater in cells from subjects with asthma.12
The collagen gel contraction assay is an established physiological in vitro model that is used to examine the mechanism of cytoskeletal reorganisation or stress fibre formation in cells such as fibroblasts13 and vascular smooth muscle cells.14 Studies using collagen gels with either smooth muscle cells from the stomach15 or aorta16 have verified that agonist induced gel contraction is actomyosin driven. The collagen gel assay has also been used to assess contraction of tracheal smooth muscle cells from bovine17 or human tissue.18 However, in these studies the gels remained attached to the casting plates17 and contraction was assessed at a single time point only, 2 h after stimulation.18 The methods used in these studies therefore did not allow for the assessment of the rate of gel contraction.
The aim of this study was to establish a refined collagen gel assay to measure the degree of contraction of human primary ASM cells in culture and to enable a comparison between contraction in ASM cells from subjects with and without asthma.
METHODS
Full details of the methods used are given in the online supplement available at http://thorax.bmj.com/supplemental.
Study population and cell culture
ASM cells were obtained from nine patients without asthma and eight with asthma (table 1) and were propagated as previously described (see fig E1 in online supplement at http://thorax.bmj.com/supplemental).19 Approval for all experiments using human lung cells was provided by the human ethics committees of the University of Sydney and the South West Sydney Area Health Service. Cells from passages 3–8 were grown to confluence using Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum and were harvested by trypsin digestion and used for experiments.
Demographic data of study patients
Collagen gel contraction assay using ASM
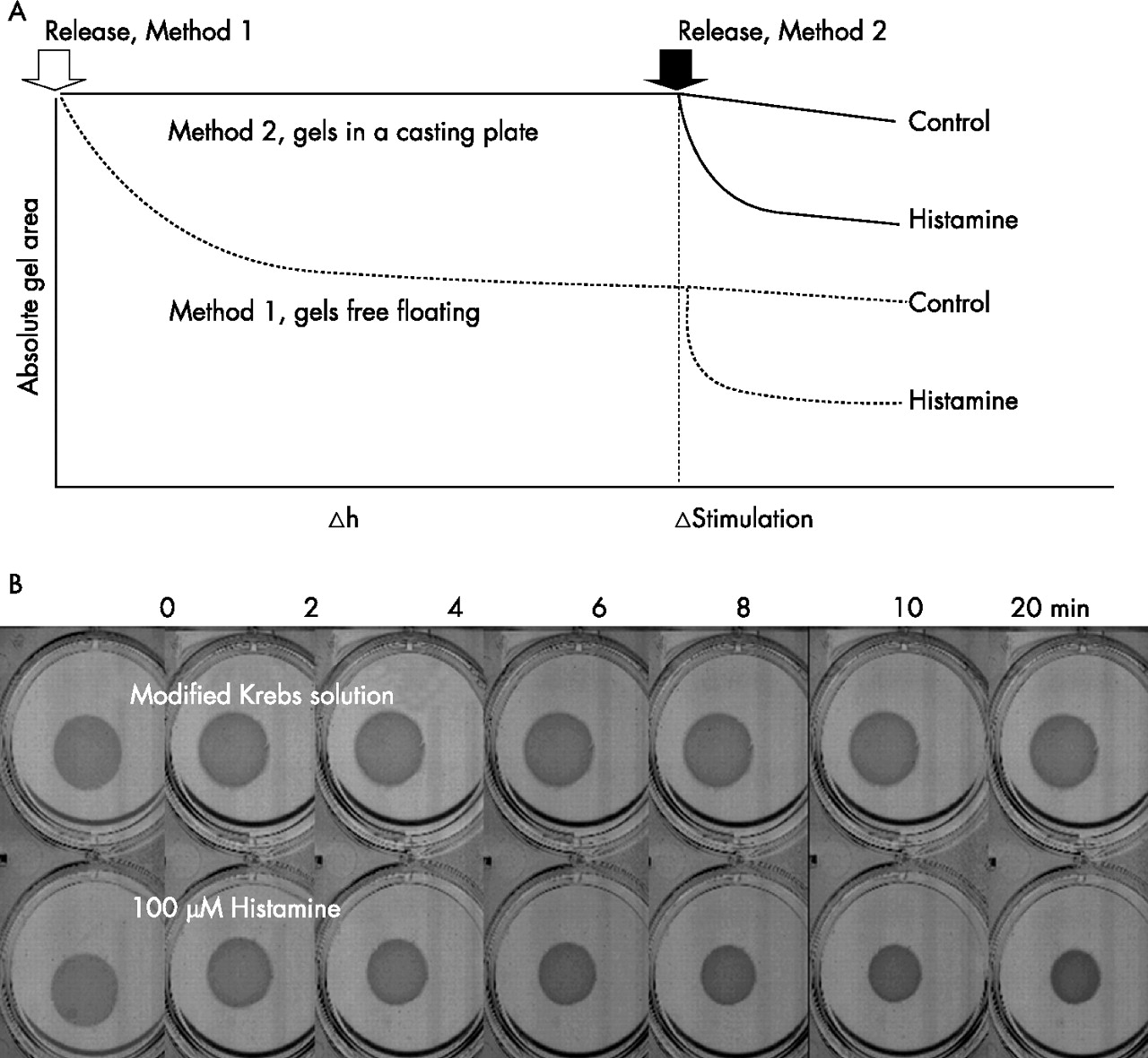
A collagen gel contraction assay was used to examine the contractile capacity of ASM cells. A collagen suspension (1.5 mg/ml) containing the ASM cells, 0.6 ml (1.5×105 cells), was cast in one well of a 24-well culture plate and allowed to polymerise (30 min, 37°C). Once polymerised, the gel was carefully detached from the culture well and transferred into a 6-well plate containing 3 ml DMEM with 0.1% bovine serum albumin (BSA). Gels containing ASM cells contracted spontaneously after detachment from the casting plates, approaching a plateau at 12 h (see fig E2 in online supplement at http://thorax.bmj.com/supplemental). To avoid the initial contraction, floating gels were equilibrated overnight in 6-well plates containing 3 ml DMEM with 0.1% BSA (37°C) before being stimulated (method 1, fig 1A). For a comparison between non-asthmatic and asthmatic ASM cells, gels stimulated immediately after detachment (method 2, fig 1A) were also examined. For method 2, collagen gels, once polymerised, were overlaid with 0.4 ml DMEM with 0.1% BSA and incubated overnight in 24-well plates before being detached.

(A) Schematic diagram representing methods 1 and 2. Arrows indicate the time of detachment for method 1 (open) and method 2 (black). The triangle denotes the time point for agonist stimulation. (B) Representative images showing the time course of gel contraction to histamine (100 µM; bottom) compared with Krebs control (top).
During gel contraction the 6-well plate was placed on a flat bed scanner (Microtek Inc, Taiwan) and was scanned automatically every 2 min over 20 min. The surface area of each gel was measured using Image J (http://rsb.info.nih.gov/ij) and gel contraction was measured at each time point as the ratio of treated gel area to untreated gel area (control) to account for any change in gel area not caused by the treatment.
Viability assay
Viability and cell number in the gels was estimated using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (Promega, NSW, Australia). The trypan blue exclusion test was also used to examine cell viability.
Western blotting
Protein expression of smooth muscle MLCK (smMLCK) was analysed by western blotting and enhanced chemiluminescence. To obtain protein from ASM cells embedded in collagen gels, the cells were harvested by digesting the collagen with collagenase for 30 min at 37°C. However, this process of cell extraction resulted in degradation of smMLCK, so the smMLCK protein content was measured for ASM cells seeded (2×104/cm2) onto collagen coated 6-well plates and left overnight. After cell lysis and centrifugation, the supernatant was collected.
Analysis of data
Data were analysed using StatView V.5.0 (SAS Institute Inc, Cary, Noroth Carolina, USA). For comparison of the two patient groups, an unpaired two-tailed Student t test or Fisher exact test was used. For the comparison of gel contraction at different time points, repeated measures two-way ANOVA with Bonferroni/Dunn correction was employed. A p value of ⩽0.05 was considered statistically significant. Data were expressed as mean (SE) values unless stated otherwise; n refers to the number of cell lines examined with each cell line being derived from a different patient.
RESULTS
Validation of histamine induced contraction of human ASM cells embedded in collagen gel
Method 1 (overnight detachment) was used to examine the effects of various contractile stimuli including carbachol (100 µM), potassium chloride (KCl) (80 mM), adenosine 5′-triphosphate (10 µM) and histamine (1–100 µM) on contraction of collagen gels that contained human ASM cells. Histamine and, to a lesser degree, adenosine 5′-triphosphate caused concentric contraction of the collagen gels within 20 min. Neither carbachol nor KCl caused significant contraction (data not shown). Human ASM cells contracted to histamine in a concentration-dependent (see fig E3A in online supplement at http://thorax.bmj.com/supplemental, n = 6, p = 0.010, two-way ANOVA) and time-dependent manner. Representative images showing the time course of histamine induced collagen gel contraction compared with untreated gels are shown in fig 1B. Contraction to histamine (1, 10, 100 µM) was significantly induced after 4 min (p<0.0001) and reached a maximum at a mean (SE) of 15.7 (1.2) min (see fig E3A in online supplement available at http://thorax.bmj.com/supplemental, n = 6). Histamine induced gel contraction was significantly inhibited when performed in the presence of the selective H1 antagonist mepyramine (1 µM) (see fig E3B in online supplement available at http://thorax.bmj.com/supplemental, n = 7, p = 0.03). Histamine (100 μM) induced gel contraction was partially reversed when treated for 20 min with formoterol (1 µM), a long-acting β2 adrenoreceptor agonist. The partial reversal of contraction was significant 16 min after the addition of formoterol (see fig E3C in online supplement available at http://thorax.bmj.com/supplemental, n = 4, p = 0.05). Pre-incubation with the physiological antagonist prostaglandin E2 significantly inhibited histamine induced gel contraction (see fig E3D in online supplement available at http://thorax.bmj.com/supplemental, n = 4, p = 0.05).
To validate the signalling pathways regulating the histamine induced contraction of ASM cells, gels were pre-incubated for 30 min with either the phospholipase C (PLC) inhibitor U73122 (10 µM), the myosin light chain kinase (MLCK) inhibitor ML-7 (30 µM), or with two different concentrations of the Rho-associated coiled coil forming kinase (ROCK) inhibitor Y27632 (3 μM and 10 μM) before the addition of histamine (100 µM). Inhibition of PLC, MLCK and ROCK significantly reduced histamine induced gel contraction (fig 2A–C, n = 3–6, p<0.05).
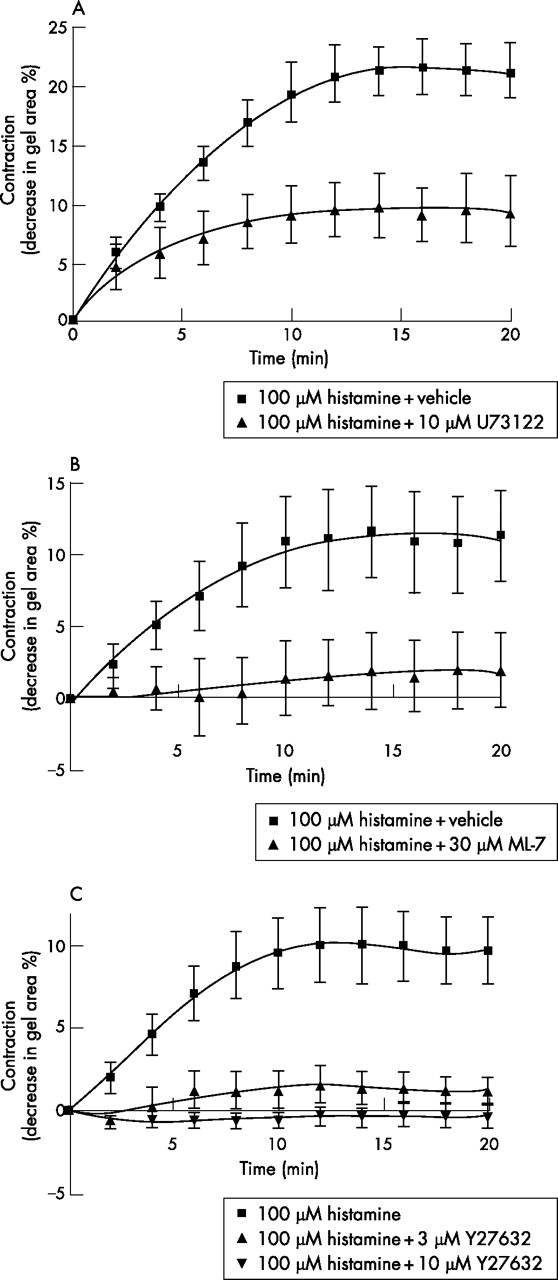
Inhibition of histamine (100 µM) induced gel contraction by (A) the phospholipase C (PLC) inhibitor U73122 (10 μM; n = 3 different patients), (B) the myosin light chain kinase (MLCK) inhibitor ML-7 (30 μM; n = 6), and (C) the Rho-associated coiled coil forming kinase (ROCK) inhibitor Y27632 (n = 5 for histamine alone and 10 μM Y27632, n = 4 for 3 μM Y27632). Contraction curves to histamine with inhibitory treatments (U73122, ML-7, 3 μM and 10 μM Y27632) were all significantly different from that of histamine alone (p⩽0.05, repeated measures two-way ANOVA).
The within experiment coefficient of variance for histamine (100 µM) induced gel contraction was measured to estimate the precision/repeatability of the experiments. The mean coefficient of variance for all time points during contraction was 1.1% when duplicate gels were compared (n = 4). In addition, cell passaging did not have a significant effect on the contraction time course curves to histamine (100 µM, n = 8; data not shown).
Comparison of histamine induced contraction in ASM cells from subjects with and without asthma using method 1
The comparison study of histamine (100 µM) induced contraction included ASM cells obtained from nine subjects without asthma and eight patients with asthma. There was no difference in sex between the two patient groups. Subjects without asthma were older than those with asthma (p<0.05, table 1).
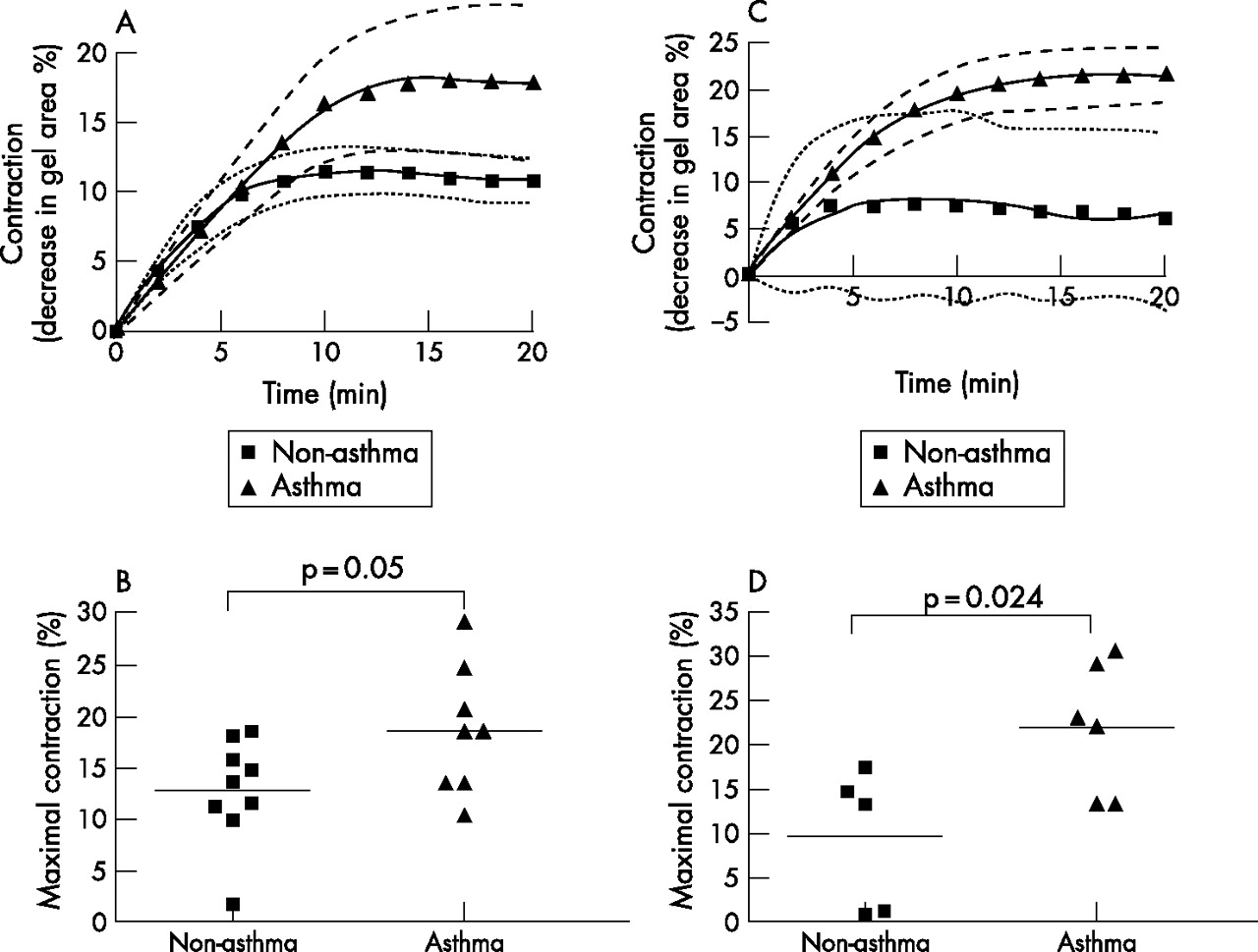
With method 1, the kinetic curves generated in response to histamine over a 20-minute period tended towards a difference between the two groups but this did not reach significance by two-way ANOVA (p = 0.073, fig 3A). However, maximal contraction was significantly greater for cells from patients with asthma than for non-asthmatic cells (19 (2)% vs 13 (2)%, p = 0.05, fig 3B). The maximum rate of contraction did not differ between those without and with asthma (2.2 (0.5)% contracted/min vs 2.1 (0.3)% contracted/min, p>0.05).

(A) Contraction curves of airway smooth muscle (ASM) cells from nine patients without asthma and eight with asthma using gels following overnight equilibration (method 1). 95% confidence intervals are indicated as dotted lines and dashed lines for non-asthmatic and asthmatic curves of best fit, respectively. (B) Maximal contraction of ASM cells from patients without and with asthma using method 1. (C) Contraction curves of ASM cells from patients without asthma (n = 5) and with asthma (n = 6) using gels detached just before stimulation (method 2). The contraction curve of ASM cells from patients with asthma using method 2 is significantly different from that of cells from non-asthmatic patients (p = 0.027). (D) Maximal contraction of ASM cells from patients without and with asthma using method 2. Each bar shows the mean value.
Comparison of histamine induced contraction in ASM cells from subjects with and without asthma using method 2
Although the baseline gel area before stimulation was not significantly different for gels embedded with ASM cells from patients without and with asthma using method 1 (1.34 (0.15) cm2 vs 1.57 (0.10) cm2, p>0.05), it was possible that equilibration of the ASM cells embedded within gels could affect contraction. We therefore examined gel contraction without the equilibration step (method 2, fig 1A). Experiments using this method were performed using ASM cells derived from five patients without asthma and six with asthma, among whom no age difference was observed (p = 0.1).
The kinetic curves to histamine contraction were significantly different when comparing ASM cells from patients without and with asthma (p = 0.027, fig 3C). Maximal contraction was also significantly greater for cells from patients with asthma than for cells from those without asthma (22 (3)% vs 10 (4)%, p = 0.024, fig 3D). Maximal contraction in method 2 was not significantly different from that in method 1 for both patient groups (p = 0.5 for patients without asthma, p = 0.1 for patients with asthma). The maximum rate of contraction was not significantly different between the two patient groups (data not shown), but the time to reach 50% of maximal contraction was significantly shorter for cells from patients without asthma than for those with asthma (1.9 (0.5) min vs 4.2 (0.5) min, p = 0.012).
The viability of cells maintained overnight in collagen gels was not significantly different between non-asthmatic patients (n = 5, 0.53 (0.12) optical density at 490 nm) and those with asthma (n = 5, 0.56 (0.12) optical density, p>0.05) as assessed by MTS assay. In all experiments cell viability as measured by trypan blue dye exclusion was >95% (data not shown).
Expression of smooth muscle MLCK
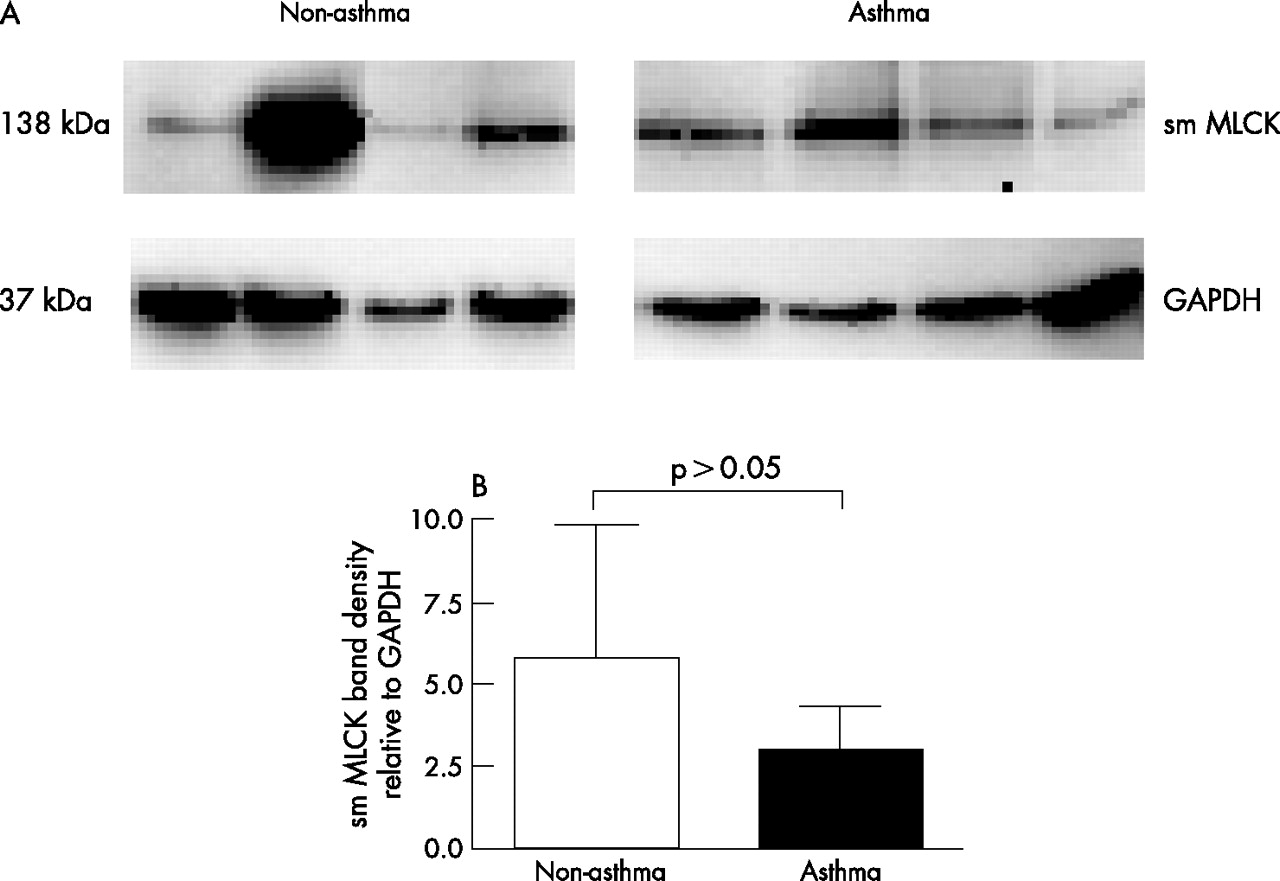
To investigate a cellular mechanism for increased contraction of asthmatic cells, we measured the amount of smMLCK in ASM cells from patients without (n = 4) and with (n = 4) asthma cultured on collagen coated plates (fig 4A). The mean (SE) ratio of the optical density for smMLCK and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression in ASM cells from patients with asthma (3.1 (1.7) densitometric units) was not significantly different from that in cells from patients without asthma (4.6 (3.6) densitometric units, p>0.05, fig 4B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Western blotting analysis for smooth muscle myosin light chain kinase (smMLCK) from airway smooth muscle (ASM) cells cultured on collagen coated plates. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level is shown as an internal control. (B) Histogram showing the densitometric ratio of smMLCK and GAPDH. Each bar shows the mean (SE) value.
DISCUSSION
This study has shown that ASM cells from patients with asthma embedded in collagen gels contracted significantly more to histamine (100 μM) than ASM cells from patients without asthma. The difference in contraction between the two groups of patients was confirmed using two different conditions: floating gels were stimulated with histamine after overnight incubation while either unattached (method 1) or attached (method 2) to casting plates (fig 1A). Histamine is an important endogenous agonist involved in allergic disease and was used as a contractile agonist in this study. This study validated that contraction to histamine of ASM cells embedded in collagen gels was mediated through well characterised signalling pathways including the histamine H1 receptor and PLC which leads to the generation of inositol 1,4,5-triphosphate and diacylglycerol. The partial inhibition of contraction by U73122 in the present study was surprising and may indicate that gel contraction was in part mediated through PLC independent mechanisms such as the CD38/cyclic ADP-ribose pathway.20 In addition, gel contraction was inhibited by the ROCK inhibitor Y27632. This finding is consistent with the recent studies which show that ROCK plays a crucial role in smooth muscle contraction involving Ca2+ sensitisation,21 myosin phosphatase inhibition22 and MLC phosphorylation.23
In the present study, only partial reversal of gel contraction was achieved following the addition of a β2 adrenoceptor agonist at the time point when contraction had reached the plateau phase. The efficacy of formoterol in the present study was poor compared with bronchial tissue which was preloaded and possessed recoil (afterload) properties. Our finding could be explained by the composition of the collagen gel which lacks the necessary viscoelastic properties for recoil to occur and/or does not readily permit re-entry of water expelled from the matrix during contraction. The poor efficacy of formoterol does not appear to result from the absence of the necessary signalling mechanisms since pretreatment with prostaglandin E2, which mediates relaxation through the cAMP cascade, caused physiological antagonism of histamine (ie, in the presence of prostaglandin E2 the response to histamine was attenuated).
The cultured ASM cell phenotype appears to differ from the contractile phenotype of ASM in the airway wall since, in our study, neither the cholinergic agonist carbachol nor the KCl membrane depolarising solution caused gel contraction. The reasons for this may be because of the downregulation of the M3 receptor and K+ channel expression, respectively.24 Despite the fact that cultured ASM cells were not responsive to the cholinergic agonist or KCl, contractility was effectively evaluated using the mast cell mediator histamine which does have its signalling pathways present in these cells. In addition, the MLCK inhibitor retarded gel contraction to a large degree which lends further support to the notion that the observed contraction is actomyosin-dependent. Despite this evidence, there still remains the possibility that gel contraction was partly mediated by reorganisation of the cytoskeleton.
We found that ASM cells from patients with asthma contracted significantly more than cells obtained from patients without asthma. This increased maximal contraction of ASM cells from patients with asthma occurred despite the use of the two different methods to assess cell contraction in collagen gels. For both methods the gels were free floating while being exposed to histamine. In method 1 the gels were equilibrated while free floating. During equilibration the gels intrinsically contracted which was thought to reflect cytoskeletal reorganisation14 rather than agonist induced actomyosin driven contraction per se. It was possible that differences in agonist induced gel contraction may have been attributed to differences in post-equilibrated gel size by altering afterloads. For this reason, a second method (method 2) was employed where the gels were left attached to the casting plates before exposure to histamine. The difference in gel contraction between the two patient groups was more significant with method 2 than with method 1. We can speculate that the condition in method 2 where gels were left attached to casting wells overnight might have pre-stressed the cells and consequently augmented the difference in response to histamine between the two patient groups. It is not likely that the rate of shortening was due to diffusion of histamine into gel, since the diffusion coefficients of small molecules such as histamine through collagen gel (1–2 mg/ml) are similar to those through water.25 Our finding that gel contraction is increased in ASM cells from patients with asthma is consistent with in vitro studies that found increased shortening for enzyme dissociated single cells from biopsy specimens from patients with asthma.12 In addition, bronchial preparations of patients with asthma contract to a greater extent than those of patients without asthma, although this has not been a consistent finding.8,26
The increased contraction of ASM cells from patients with asthma was not attributed either to greater cell number or cell size. ASM cells from patients with asthma have been shown to proliferate faster.19 However, overnight incubation of ASM cells in a collagen gel did not cause a difference in cell number between patients with and without asthma as assessed using the MTS assay. Although we did not measure average cell length for the two patient groups, cell size as assessed by flow cytometry was not different between the two groups.27
There was a significant difference in age between the two patient groups when using method 1 but not for method 2. To date, no study using human tissue or cells has examined the effect of age on the development of stress in smooth muscle. Animal studies have found age related decreases in contraction but these differences occurred only when comparing mature with immature animals.28,29 In a study of cell proliferation rates, human cultured skin fibroblasts from 150 patients showed no effect of age.30 We admit that it is conceivable that the differences in contraction could be partly mediated by an age dependent effect. However, even if the age of the patient could affect the degree of contraction, the difference in age was not significant for method 2, which gave a more significant difference in contraction between the two patient groups.
To explain the greater maximal airway narrowing in asthma, several potential factors that modulate ASM contraction have previously been investigated. In addition to geometric and dynamic factors such as increased airway wall area,31 decreased parenchymal tethering,32 decreased fluctuating loads that perturb myosin binding during breathing,33 several inflammatory cytokines such as tumour necrosis factor α and interleukin-1β could increase contraction of ASM cells from patients with asthma by mobilising intracellular Ca2+ possibly through upregulation of CD38.20 Transforming growth factor β increases the expression of α-smooth muscle actin, the formation of actomyosin filament and cell shortening to acetylcholine.34 Although the extent to which the effect of such an in vivo milieu is retained in cultured cells is unknown, there is the possibility that ASM cells from patients with asthma are intrinsically different from cells from patients without asthma. For example, a recent study using ASM cells cultured from two different rat strains has shown that the increased airway hyperresponsiveness exhibited in the Fisher rat in vivo was reflected in the development of a greater agonist induced tension in ASM cells cultured from the same rat strain.35 This finding suggests that differences in cells in culture can reflect differences exhibited in vivo.
In this study the smMLCK protein content was not significantly different between ASM cells from patients with and without asthma cultured on collagen coated plates. Cells were grown on collagen coated coverslips instead of collagen gels to circumvent the observation in a preliminary study that showed degradation of smMLCK protein. It is possible that degradation occurred as a result of the cell harvesting process which involved incubating the collagen gel for 30 min at 37°C in a solution containing collagenase. The finding of no difference in smMLCK content is consistent with the lack of difference in the maximum rate of contraction between the two patient groups. However, our finding is not consistent with a previous study that showed an increase in smMLCK protein content in human airway tissue that was sensitised to allergens,36 nor does it agree with increased mRNA expression of the smMLCK reported in ASM cells from patients with asthma.12 However, the issue is controversial as one study using bronchi from patients with asthma showed augmented immunohistochemical expression of MLCK,3 while another using laser capture microdissection of ASM and real-time PCR did not.4 We cannot explain the smMLCK results in the present study, but it should be taken into consideration that the expression of contractile proteins might be different when ASM cells are cultured in two-dimensional rather than three-dimensional conditions.37
In conclusion, we have shown that ASM cells embedded in collagen gels can be used as a model of agonist induced actomyosin driven ASM cell contraction. Greater contraction of ASM cells from patients with asthma than from patients without asthma may account for the greater maximal airway narrowing which occurs in asthma.
Acknowledgments
The authors acknowledge the collaborative effort of the cardiopulmonary transplant team and the pathologists at St Vincent’s Hospital, Sydney and the thoracic physicians and pathologists at Royal Prince Alfred Hospital, Concord General Hospital, Strathfield Private Hospital and Rhodes Pathology, Sydney; Drs Gregory King and Melissa Baraket at the Woolcock Institute of Medical Research for supplying the asthmatic biopsies; Joanne Thompson and Pablo Britos for excellent technical assistance; and Aneal Chandra for writing a custom program for autoscanning.
REFERENCES
Supplementary materials
Files in this Data Supplement:
Footnotes
-
Published Online First 5 April 2007
-
This work was supported by the National Health and Medical Research Council of Australia (NH&MRC).
-
Competing interests: None.
Linked Articles
- Airwaves