Article Text

Abstract
Background: The role of anti-granulocyte-macrophage colony stimulating factor (GM-CSF) antibodies as a diagnostic marker in idiopathic pulmonary alveolar proteinosis (iPAP) remains unclear.
Methods: Anti-GM-CSF antibodies were detected in blood and bronchoalveolar lavage fluid (BAL) fluid in 13 patients with iPAP. Three patients with secondary PAP, 35 with other pulmonary disorders, and 10 subjects without lung lesions acted as controls. Blood samples only were obtained from 30 healthy medical personnel. Anti-GM-CSF antibodies were detected using immunoblotting and measured semi-quantitatively by serial dilution or concentration methods. The relationship between antibodies and reported severity indicators for iPAP was analysed.
Results: Anti-GM-CSF antibodies could be detected in both blood and BAL fluid samples in 12 of 13 iPAP patients and were undetectable in blood and/or BAL fluid from the other subjects studied. BAL fluid levels of anti-GM-CSF antibodies were highly correlated with the severity indicators for iPAP, including serum lactate dehydrogenase (LDH) levels, arterial oxygen tension, alveolar-arterial oxygen tension difference, (AaPo2), lung carbon monoxide transfer factor, and some lesion scores on chest radiographs and computed tomographic scans. In contrast, blood anti-GM-CSF antibodies were not significantly correlated with the severity indicators evaluated. In addition, patients with iPAP who required subsequent therapeutic lung lavage had significantly higher values of serum LDH, AaPO2, and BAL fluid anti-GM-CSF antibodies, and significantly lower values of Pao2.
Conclusions: In addition to serum LDH levels, Pao2 and AaPo2, BAL fluid levels of anti-GM-CSF antibodies might reflect disease severity in patients with iPAP and predict the need for subsequent therapeutic lung lavage. These findings may expand the role of anti-GM-CSF antibodies in iPAP.
- AaPo2, alveolar-arterial oxygen tension difference
- BAL, bronchoalveolar lavage
- GM-CSF, granulocyte-macrophage colony stimulating factor
- iPAP, idiopathic pulmonary alveolar proteinosis
- LDH, lactate dehydrogenase
- Pao2, arterial oxygen tension
- Tlco, carbon monoxide transfer factor
- anti-granulocyte-macrophage colony stimulating factor antibodies
- granulocyte-macrophage colony stimulating factor
- lactate dehydrogenase
- pulmonary alveolar proteinosis
- pulmonary function
Statistics from Altmetric.com
- AaPo2, alveolar-arterial oxygen tension difference
- BAL, bronchoalveolar lavage
- GM-CSF, granulocyte-macrophage colony stimulating factor
- iPAP, idiopathic pulmonary alveolar proteinosis
- LDH, lactate dehydrogenase
- Pao2, arterial oxygen tension
- Tlco, carbon monoxide transfer factor
- anti-granulocyte-macrophage colony stimulating factor antibodies
- granulocyte-macrophage colony stimulating factor
- lactate dehydrogenase
- pulmonary alveolar proteinosis
- pulmonary function
Pulmonary alveolar proteinosis (PAP) is a rare disease characterised by accumulation of surfactant within the alveoli and terminal airways. Two types of PAP can be recognised—congenital and acquired—while acquired PAP can be divided further into idiopathic and secondary forms. Clinically, in more than 90% of patients with PAP the disease is of unknown aetiology—that is, idiopathic PAP (iPAP).1,2
Macrophage dysfunction has been thought to be responsible for the surfactant accumulation in iPAP, but the exact aetiology and pathogenesis remain obscure.1–,3 In 1994 investigations on experimental haematology unexpectedly established a link between granulocyte-macrophage colony stimulating factor (GM-CSF) and surfactant homeostasis.4,5 Without changes in basal haematopoiesis, mice deficient in either GM-CSF or its receptor developed pulmonary lesions histologically similar to PAP.4–,6 Further studies showed that local availability of GM-CSF was sufficient to correct surfactant accumulation in GM-CSF knockout mice, either by aerosolised administration7 or selective expression of GM-CSF in respiratory epithelial cells.8 Exogenous GM-CSF treatment was subsequently applied to patients with iPAP with a response rate of 40–50%.1,2,9–,11
Unlike the animal model, the defects in gene expression of GM-CSF or its receptor have not yet been found in human iPAP.1,12,13 There were no intrinsic cellular defects in synthesising and secreting GM-CSF,14 but the bioavailability of GM-CSF appeared to be blocked by neutralising antibodies.14,15 High levels of neutralising antibodies against GM-CSF could be detected in the blood and bronchoalveolar lavage (BAL) fluid from patients with iPAP, but not in those of patients with congenital or secondary PAP, patients with other pulmonary diseases, or in healthy subjects, suggesting that iPAP may be an autoimmune disease.1–3,15 By blocking the binding of GM-CSF to its receptor and thereby inhibiting the differentiation and function of macrophages, anti-GM-CSF antibodies are considered to be a causal factor of iPAP.16 Nevertheless, the clinical significance of anti-GM-CSF antibodies in iPAP remains obscure, as evidenced by mixed results of anti-GM-CSF antibodies in predicting the treatment response to GM-CSF and lack of correlation between blood anti-GM-CSF antibodies and reported severity markers for iPAP.11,17 An increase in arterial oxygen tension (Pao2) accompanied by a decrease in anti-GM-CSF antibodies in blood and/or BAL fluid was reported recently in three patients with iPAP after treatment with inhaled GM-CSF.18
Based on these findings, the clinical relevance of anti-GM-CSF antibodies in iPAP deserves further study. We measured anti-GM-CSF antibodies in paired specimens of blood and BAL fluid from patients with iPAP and investigated their relation to severity markers for iPAP including serum lactate dehydrogenase (LDH), pulmonary function parameters, arterial blood gases, and radiological findings.1–3,11,19
METHODS
Patients with iPAP
Thirteen consecutive iPAP patients diagnosed by cytological examination of BAL fluid20 and pathological examination of the lung tissues at Taipei Veterans General Hospital were enrolled in the study. The aetiologies reported to be associated with secondary PAP were not found in these patients. All patients studied are alive at the time of manuscript preparation. One of the patients had diabetes mellitus and hypertension treated with hypoglycaemic and antihypertensive agents. The others denied any major medical illnesses. The Institutional Review Board of Taipei Veterans General Hospital approved the study and informed consent was obtained from the patients for the study of blood and BAL fluid samples.
All iPAP patients were followed up for more than 1 year except for the one diagnosed in March 2005. The disease improved spontaneously in three and remained stable in five. Therapeutic lung lavage was required in the remaining five patients. The criteria for therapeutic lung lavage in this study were dyspnoea with restriction of daily activity and/or occupation or Pao2 <8 kPa on room air. Nearly complete resolution was observed in one patient after three cycles of therapeutic whole lung lavage. Repeated therapeutic lung lavage was necessary for one other patient. Because of poor response and intolerance to therapeutic whole lung lavage, therapeutic bronchoscopic lobar/segmental lavage was adopted subsequently in this patient.21 The remaining three received one cycle of bronchoscopic lobar/segmental lavage with remarkable improvement as evidenced by follow up imaging studies and pulmonary function testing. None of the patients received GM-CSF therapy.
Serum LDH, pulmonary function testing, chest radiography, and high resolution computed tomography (HRCT) of the chest were performed before diagnostic BAL at an interval of less than 3 days. Only BAL fluid samples obtained from diagnostic BAL at the time of initial diagnosis were included for analysis.
Pulmonary function testing
Pulmonary function testing including spirometry, plethysmography, carbon monoxide lung transfer factor (Tlco), and analyses of arterial blood gases were performed in all patients. Blood samples for pH, Pao2 and Paco2 (arterial CO2 tension) values were analysed at room air using ABL III (Radiometer, Copenhagen, Denmark). Pao2 (alveolar oxygen tension) is calculated by the following equation. Pao2 = (barometric pressure − 47) × Fio2 − Paco2/R. R, an exchange ratio, is assumed as 0.8 in this study. The alveolar−arterial Po2 difference (AaPO2) is calculated by subtracting Pao2 from Pao2.
Image scoring
Frontal chest radiographs and HRCT scans of the chest were graded according to the visual scoring system proposed by Lee et al.19 Scores were determined for degree, extent, and severity of lung opacity. The opacity score was evaluated by means of a three-point scale as follows: mild ground glass opacity = 1; moderate ground glass opacity = 2; consolidation opacity = 3. The “ground glass opacity” refers to the presence of increased lung opacity associated with partial obscuring of normal vascular structures. The extent of lung opacity was estimated by means of four-point scale: opacity involving <25% of hemithorax = 1; ⩾25 to <50% = 2; ⩾50 to <75% = 3; ⩾75% = 4. The severity score was calculated by multiplying the opacity and extent scores. The final values for these scores were the sum of each hemithorax. The chest radiographs were read and interpreted independently by two chest physicians (S-CC and F-CL) who were blind to the clinical data and results of pulmonary function testing. The mean values obtained from the two readers were used for analysis.
The HRCT images were photographed at window levels appropriately for demonstrating the pulmonary parenchyma, and were read and interpreted by two chest physicians (S-CC and F-CL) and one chest radiologist (M-SC) in a blind fashion. Every individual scan was evaluated for the degree, extent and severity of lung opacity, and the extent of reticulation. The degree and extent of opacity were scored using the same scales as described above. The severity score was calculated by multiplying the opacity and extent scores. The extent of reticulation was measured as the extent of lung opacity. The score for each lung was the average of all HRCT slices, and the sum of each hemithorax became the final. The mean values obtained from the three readers were used for analysis.
BAL fluid and blood samples
The fibreoptic bronchoscope (Model BF 20 or P20; Olympus, Tokyo, Japan) was wedged into the orifice of lobar or segmental bronchus of the right middle or lingular division, or other appropriate location. Diagnostic BAL was performed before transbronchial lung biopsy using three aliquots of a 50 ml sterile isotonic sodium chloride solution. The aspirates were pooled into a siliconised container and kept on ice during transport. A 10 ml sample of venous blood was collected immediately after BAL. The supernatants of blood and BAL fluid were frozen and stored at −70°C until analysed.
Measurement of GM-CSF
The levels of GM-CSF in blood and BAL fluid were measured by a commercially available enzyme linked immunosorbent assay (ELISA) kit (R&D System, Minneapolis, MN, USA) with a lower limit of 3 pg/ml.
Measurement of anti-GM-CSF antibodies
The levels of anti-GM-CSF antibodies were detected and measured by immunoblotting. In brief, human recombinant GM-CSF (E coli derived; molecular mass 14 kD; Calbiochem, Affiliate of Merck KGaA, Darmstadt, Germany) was subjected to 8% polyacrylamide gel electrophoresis under reducing condition with 0.1 μg rhGM-CSF loaded in each lane. The proteins were then transferred to polyvinylidene fluoride (PVDF) membrane by diffusion blotting.22 With appropriate blocking, incubation and washes, horseradish peroxidase-conjugated anti-human IgG antibodies (Jackson Immuno Research Laboratory, PA, USA) was added and the immunoreactive bands were developed by 3.3-diaminobenzidine, H2O2 and NiCl2 enhancement.
For the blood and BAL fluid samples with positive results, the concentrations of anti-GM-CSF antibodies were determined semi-quantitatively by serial dilution. In case of negative results, the BAL fluid samples were concentrated for further measurement.
Control groups
To serve as disease controls, paired specimens of blood and BAL fluid were obtained from three patients with secondary PAP and 35 patients with other pulmonary diseases including collagen vascular diseases in 12, cytomegalovirus pneumonitis in 12, idiopathic pulmonary fibrosis in six, and sarcoidosis in five patients. The underlying diseases of secondary PAP were acute myeloid leukaemia in two patients and renal transplantation in one. Blood and BAL fluid were obtained from 10 patients without pulmonary lesions to serve as lung controls. Peripheral blood only was obtained from 30 healthy hospital personnel to serve as normal controls.
Statistical analysis
Data were expressed as median (range). Paired data comparisons were performed using a Wilcoxon signed rank test. Specific comparisons of data between two groups were made using the Mann-Whitney U test. The correlations between variables were determined by Spearman rank correlation coefficients. Significance was defined as p<0.05. Statistical analysis was performed using SPSS Version 10 (SPSS, Chicago, IL, USA).
RESULTS
From January 1995 to March 2005, 13 consecutive patients with iPAP of varying degrees of severity were enrolled in the study. Their demographic characteristics and clinical data are summarised in table 1⇓.
Clinical characteristics of patients with idiopathic pulmonary alveolar proteinosis
Correlations between severity markers for iPAP
The relationships between the severity indicators for iPAP—including serum LDH, Pao2 and AaPo2, results of pulmonary function testing, and image scores—are given in table 2⇓. Patients with iPAP had varying degrees of restrictive ventilatory defect. Gas exchange was impaired, as evidenced by a reduction in Tlco or Pao2, or a widening of AaPo2. The levels of serum LDH were abnormally high in 11 of the 13 cases. The values of Pao2 and AaPo2 correlated significantly with those of serum LDH (r = −0.69, p = 0.010, and r = 0.60, p = 0.029, respectively). The ventilatory function data did not correlate significantly with those of serum LDH, Pao2 and AaPo2, but there was a negative correlation between Tlco and AaPo2.
Relation between clinical, radiological, and physiological parameters in patients with idiopathic pulmonary alveolar proteinosis (iPAP)
The common findings of iPAP on the chest radiographs were bilateral airspace ground glass opacity and/or consolidation with relatively symmetrical distribution. The severity score on the chest radiographs was highly correlated with serum LDH, Pao2, and AaPo2 (table 2⇑), and the extent score on the chest radiographs was highly correlated with serum LDH and AaPo2. A negative correlation was found between total lung capacity (TLC) and all lesion scores (opacity score, r = −0.65, p = 0.017; extent score, r = −0.64, p = 0.020; severity score, r = −0.69, p = 0.009) on the chest radiographs. The severity score on the chest radiographs correlated negatively (r = −0.55, p = 0.049) with forced vital capacity (FVC). Tlco was not highly correlated with lesion scores on the chest radiographs.
Characteristic findings of iPAP on thoracic HRCT scans revealed patchy ground glass opacity with varying degrees of intralobular and interlobular septal thickening. The extent, severity and reticulation scores of the HRCT scans were highly correlated with those of serum LDH, Pao2 and AaPo2 (table 2⇑). All lesion scores on the HRCT scans were highly correlated with TLC (opacity score: r = −0.82, p = 0.001; extent score: r = −0.69, p = 0.009; severity score: r = −0.77, p = 0.002; reticulation score: r = −0.84, p<0.001). The severity and reticulation scores on the HRCT scans were negatively correlated with FVC (r = −0.62, p = 0.025 and r = −0.57, p = 0.042, respectively). Similarly, the lesion scores on the HRCT scans were not highly correlated with Tlco.
GM-CSF in iPAP patients and control groups
In patients with iPAP there was no significant difference in blood and BAL fluid levels of GM-CSF. Compared with those with secondary PAP, patients with iPAP had significantly lower blood and BAL fluid levels of GM-CSF (p = 0.025, respectively, fig 1⇓). The blood levels of GM-CSF were significantly higher in iPAP patients than in normal controls, lung controls, and in patients with cytomegalovirus pneumonitis. In contrast, there was no significant difference in BAL fluid levels of GM-CSF in patients with iPAP, those with other pulmonary disease, and lung controls.

Box and whisker plots for the levels of GM-CSF in (A) blood and (B) BAL fluid. The box represents the interquartile range that contains 50% of the values and the whiskers extending from the box represent the highest and lowest values. iPAP, idiopathic pulmonary alveolar proteinosis; IPF, idiopathic pulmonary fibrosis; CVD, collagen vascular disease; CMV, cytomegalovirus pneumonitis; NC, normal controls.
Anti-GM-CSF antibodies in iPAP patients and control groups
Anti-GM-CSF antibodies could be detected in both blood and BAL fluid samples in 12 of the 13 patients with iPAP. By contrast, anti-GM-CSF antibodies could not be detected in blood or BAL fluid samples obtained from disease controls, lung controls, and normal controls. Accordingly, the sensitivity and specificity of anti-GM-CSF antibodies determined by immunoblotting in aiding a diagnosis of iPAP were 92% and 100%, respectively.
For samples with positive results, the levels of anti-GM-CSF antibodies were determined semi-quantitatively by serial dilution. For samples with negative results, BAL fluid was concentrated for further evaluation. In subjects with undetectable antibodies the levels of blood and BAL fluid antibodies were defined arbitrarily as 1:1 (1) and 50:1 (0.02), respectively. The distribution of anti-GM-CSF antibodies in patients with iPAP and the various control groups are shown in fig 2⇓.

Anti-GM-CSF antibodies measured by immunoblotting in (A) blood and (B) BAL fluid. Median values and ranges are given for patients with iPAP. Anti-GM-CSF antibodies, anti-granulocyte-macrophage colony stimulating factor antibodies; BALF, bronchoalveolar lavage fluid; iPAP, idiopathic pulmonary alveolar proteinosis; LC, lung controls; IPF, idiopathic pulmonary fibrosis; CVD, collagen vascular disease; CMV, cytomegalovirus pneumonitis; NC, normal controls.
Correlations between anti-GM-CSF antibodies and severity markers for iPAP
The relationship between anti-GM-CSF antibodies and severity markers for iPAP was analysed and the results are shown in table 3⇓. The levels of blood anti-GM-CSF antibodies did not correlate with those of the clinical parameters evaluated. In contrast, the levels of BAL fluid anti-GM-CSF antibodies were significantly correlated with those of serum LDH, Pao2, AaPo2, Tlco, opacity score on the chest radiographs, and the extent and reticulation scores on the HRCT scans. In terms of evaluating the disease severity of iPAP, local or BAL fluid anti-GM-CSF antibodies were superior to those in the circulation.
Relationship between anti-GM-CSF antibodies and severity indicators for iPAP
Anti-GM-CSF antibody levels and the need for therapeutic lung lavage
To explore further the clinical significance of anti-GM-CSF antibodies in iPAP, 13 patients were divided into subgroups based on the need for subsequent therapeutic lung lavage. The patients who required therapeutic lung lavage tended to have higher values of serum LDH, AaPo2, and HRCT scores, and lower values of Pao2, FEV1, FVC, TLC, and Tlco (table 4⇓). The differences in serum LDH, Pao2, AaPo2, and extent and reticulation scores on the HRCT scans were statistically significant. Furthermore, these patients had significantly higher titres of anti-GM-CSF antibodies in the BAL fluid at the time of diagnosis of iPAP.
Comparisons between iPAP patients divided into groups based on the need for therapeutic lung lavage
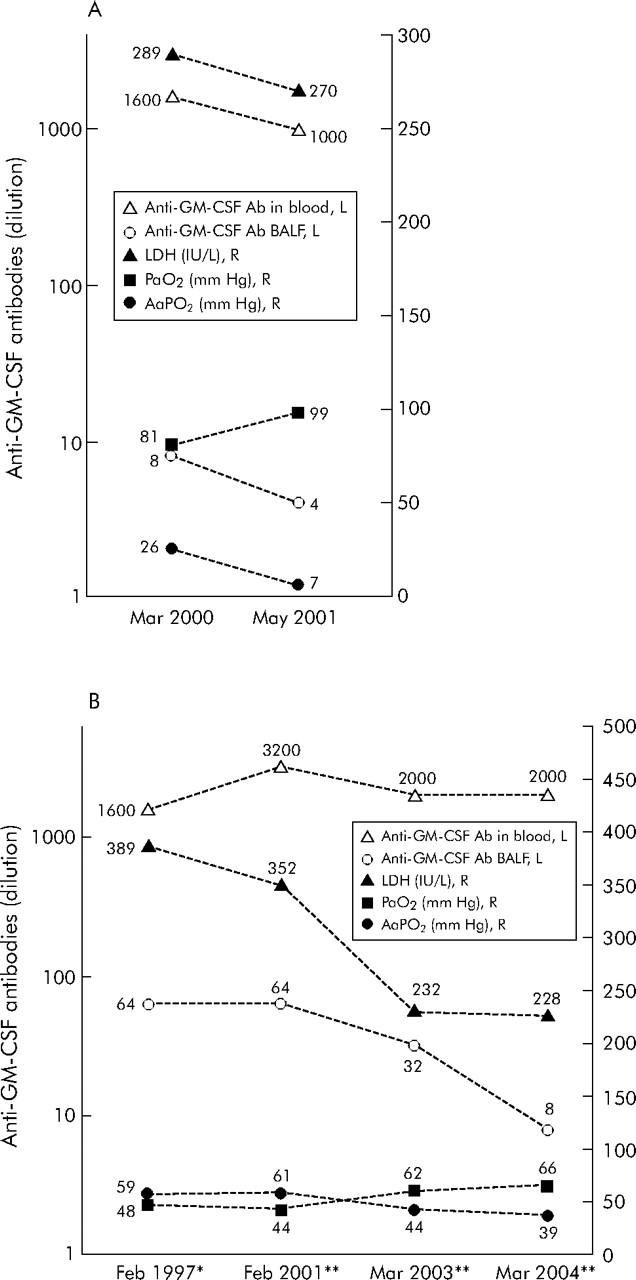
Serial changes in anti-GM-CSF antibodies in two iPAP patients with different clinical course
Serial changes in anti-GM-CSF antibodies and severity markers for iPAP in two patients are shown in fig 3⇓. In the patient with spontaneous improvement, the improvement in serum LDH, AaPo2 and Pao2 were found in association with a decrease in anti-GM-CSF antibodies in blood and BAL fluid. In the patient who required repeated therapeutic lung lavage, the changes in BAL fluid and blood anti-GM-CSF antibodies measured before each cycle of therapeutic lung lavage were relatively parallel to those of the severity markers.
{kind=link}
{kind=link}
{kind=link}
Serial data of anti-GM-CSF antibodies in blood and BAL fluid and severity indicators in two patients with iPAP with a different clinical course: (A) a patient who improved spontaneously and (B) a patient who required repeated therapeutic lung lavage. In (B), all the data were obtained before each cycle of therapeutic lung lavage. *Treated with therapeutic whole lung lavage. **Treated with therapeutic bronchoscopic lobar/segmental lavage. Anti-GM-CSF antibodies, anti-granulocyte-macrophage colony stimulating factor antibodies; BALF, bronchoalveolar lavage fluid; iPAP, idiopathic pulmonary alveolar proteinosis; LDH, lactate dehydrogenase; Pao2, arterial oxygen tension; AaPo2, alveolar-arterial oxygen tension difference.
DISCUSSION
This is the first study to demonstrate a relation between BAL fluid levels of anti-GM-CSF antibodies and severity indicators for iPAP, including serum LDH, Tlco, Pao2, AaPo2, and some lesion scores on chest radiographs and HRCT scans. In terms of evaluating the severity of iPAP, BAL fluid anti-GM-CSF antibodies were superior to those in the circulation. BAL fluid levels of anti-GM-CSF antibodies might predict the need for therapeutic lung lavage in iPAP patients. In addition, the present study supports the view that the presence of anti-GM-CSF antibodies in blood and/or BAL fluid could serve as a useful diagnostic marker for iPAP. These findings may expand the role of anti-GM-CSF antibodies in iPAP.
Several studies have indicated that circulating anti-GM-CSF antibodies could serve as a serological marker for diagnosing iPAP, and iPAP is regarded as an autoimmune disease accordingly.1–3,15,23,24 The present study indicated that, using immunoblotting, anti-GM-CSF antibodies could be detected in both blood and BAL fluid in 12 of 13 patients with iPAP. Furthermore, anti-GM-CSF antibodies could not be detected in blood and/or BAL fluid from patients with secondary PAP and other pulmonary diseases and from subjects without lung diseases (lung controls) and normal controls. These findings are consistent with previous reports23,24 that anti-GM-CSF antibodies are highly specific for iPAP. However, one of our patients who had undetectable anti-GM-CSF antibodies detected by immunoblotting in both blood and BALF did not fit into this pathophysiological group. Similarly, no significant increase in anti-GM-CSF antibodies was reported in two of seven patients with iPAP in a recent study.25 The reasons why anti-GM-CSF antibodies are not detected or are not significantly increased in blood and/or BAL fluid of some patients with iPAP remain unknown. There are several possibilities. Firstly, the methods used to detect anti-GM-CSF antibodies are not sufficiently sensitive; secondly, iPAP may be a syndrome rather than a disease; and finally, as in collagen vascular diseases, autoantibody specific for a given collagen vascular disease is not exclusively present. Further studies are required to clarify these issues.
To further verify the accuracy of immunoblotting, an in-house ELISA assay was performed as described by Schoch et al26 with some modification. Good correlation was generally observed between the levels of anti-GM-CSF antibodies measured by immunoblotting and ELISA methods in both blood (r = 0.77, p = 0.002) and BAL fluid (r = 0.70, p = 0.008). Compared with those of the control groups, significantly higher levels of anti-GM-CSF antibodies were detected in blood and BAL fluid samples in 12 of 13 patents with iPAP. The lowest values were found in the one patient who had undetectable anti-GM-CSF antibodies in both blood and BAL fluid using immunoblotting. Further details are provided in the online supplement available at http://www.thoraxjnl.com/supplemental.
Except for serving as a diagnostic marker for iPAP, the role of anti-GM-CSF antibodies in the aetiology and pathogenesis, monitoring disease activity, reflecting disease severity, predicting and/or monitoring treatment response, and in predicting outcome of the patients remains unknown. A study by Seymour et al11 reported that the circulating anti-GM-CSF antibodies did not significantly correlate with the severity indicators for iPAP. In agreement with this report,11 we found that blood levels of anti-GM-CSF antibodies did not correlate closely with AaPo2, Pao2, serum LDH levels, and lesion scores on chest radiographs and HRCT scans (table 3⇑). By contrast, the BAL fluid values of anti-GM-CSF antibodies were significantly correlated with those of serum LDH, Pao2, AaPo2, Tlco, and some lesion scores on chest radiographs and HRCT scans. Thus, BAL fluid levels of anti-GM-CSF antibodies were better than blood levels in reflecting disease severity of iPAP.
To further explore the clinical values of anti-GM-CSF antibodies in iPAP, the patients were divided into subgroups based on the need for therapeutic lung lavage (table 4⇑). Significantly higher levels of serum LDH, AaPo2 and BAL fluid anti-GM-CSF antibodies, a lower Pao2, and more severe lung lesions on the chest CT scan were found in iPAP patients who required therapeutic lung lavage. The BAL fluid levels of anti-GM-CSF antibodies measured at the time of diagnosis may therefore be of value in predicting the need for subsequent therapeutic lung lavage in iPAP patients.
There are some limitations in this study. The cases studied were limited because iPAP is a rare disease. The power of the test would be increased up to ⩾0.8 to detect a significant difference if the number of cases was doubled. However, the results can be clinically relevant since all studied patients came from the same institute and all samples were collected and processed with the same protocol. This may decrease or avoid the inherent bias caused by different methods used in collecting and/or processing samples as occurs in multicentre studies. Further studies with a larger population are needed to verify these issues.
In summary, the current study supports the view that the presence of anti-GM-CSF antibodies in blood or BAL fluid can serve as a diagnostic marker for iPAP. Furthermore, anti-GM-CSF antibodies in the BAL fluid might reflect disease severity and predict the need for subsequent therapeutic lung lavage. These findings may expand the role for anti-GM-CSF antibodies in patients with iPAP.
REFERENCES
Supplementary materials
HTML Page - index.htslp
Files in this Data Supplement:
Footnotes
Published Online First 3 March 2006
This work was supported by grants from the Taipei Veterans General Hospital (TVGH91-323 and TVGH93-288)
Competing interests: none.