Article Text

Abstract
Airway inflammation is central to the pathogenesis of both airway remodelling and parenchymal destruction in chronic obstructive pulmonary disease (COPD). Neutrophils, macrophages, and CD8+ T lymphocytes have been implicated in a number of studies, but a detailed profile of disease-phenotype specific inflammation has yet to emerge. The heterogeneity of the disease has hindered data interpretation while extrapolation of the results of relatively non-invasive studies to the actual pathology found in the distal lung is difficult. Moreover, prominent studies have had frequently conflicting results. Further investigations are needed to marry the different clinical phenotypes of COPD to their respective inflammatory profiles in the airways and thus improve our understanding of the pathogenesis of the disease as a whole.
- BAL, bronchoalveolar lavage
- COPD, chronic obstructive pulmonary disease
- ECP, eosinophilic cationic protein
- EGFR, epidermal growth factor receptor
- FEV1, forced expiratory volume in 1 second
- IFN-γ, interferon γ
- IL, interleukin
- LTB4, leukotriene B4
- MMP, matrix metalloproteinase
- MPO, myeloperoxidase
- NE, neutrophil elastase
- NK, natural killer
- TNF-α, tumour necrosis factor α
- smoking
- chronic obstructive pulmonary disease
- airway inflammation
Statistics from Altmetric.com
- BAL, bronchoalveolar lavage
- COPD, chronic obstructive pulmonary disease
- ECP, eosinophilic cationic protein
- EGFR, epidermal growth factor receptor
- FEV1, forced expiratory volume in 1 second
- IFN-γ, interferon γ
- IL, interleukin
- LTB4, leukotriene B4
- MMP, matrix metalloproteinase
- MPO, myeloperoxidase
- NE, neutrophil elastase
- NK, natural killer
- TNF-α, tumour necrosis factor α
Asthma has been the dominant focus of airways research interest, at least until recent times. A multitude of studies have examined its tapestry of airway inflammation and remodelling. Sophisticated theories have resulted, encompassing environmental exposures, epithelial repair, and epithelial-mesenchymal signalling.1 Therapeutic successes have followed, encouraging more interest and further investment. Chronic obstructive airways disease (COPD), on the other hand, has suffered from the unflattering contrast. COPD is generally considered a self-inflicted condition that results, in the majority, from years of smoking and the only effective treatment to date is long term oxygen therapy. Its profile of airways inflammation is, in retrospect, unsurprising. Neutrophils, macrophages, and CD8+ T lymphocytes are, after all, predictable responders to a sustained noxious insult. However, in recent years something of a renaissance has occurred. COPD inflammation research has burgeoned, new treatments are being tested,2 and interests are focused now not so much on the cell types but more on the “whys and wherefores” of their actions in COPD.
Historically, the link between airways inflammation and lung pathology in smokers had long been suspected. In the 1950s a post-mortem study of centrilobular emphysema revealed “bronchiolitis” of the airways leading into emphysematous parenchyma.3 Further studies confirmed not only that peripheral airways inflammation is associated with remodelling and destruction,4–8 but also that its severity increases in parallel with emphysema severity.8–10 They also revealed that bronchiolar inflammation and emphysema share the same anatomical distribution,9,11 that small airways disease temporally precedes emphysema,4 and that bronchiolar inflammation severity correlates with functional impairment.5,8,12 It was therefore hypothesised that centrilobular emphysema results from an inflammatory reaction that spreads centrifugally from the bronchioles to involve the parenchyma.9
Animal model studies and in vitro experiments have explored the roles of specific inflammatory cells in COPD, while bronchoscopy, sputum induction and lung resection studies have described airways inflammation in vivo. All have helped to confirm dominant positions in the inflammatory hierarchy for neutrophils,13–15 macrophages,16–18 and CD8+ T lymphocytes.19–21 Other inflammatory cells such as mast cells,16 eosinophils,22 and natural killer cells16 have been credited with less importance.
KEY INFLAMMATORY CELLS IN COPD
Neutrophils
Neutrophils are front line defensive cells of the immune system and a source of reactive oxygen metabolites, inflammatory cytokines, lipid mediators, antibacterial peptides, and tissue damaging enzymes.23–25 They are strongly implicated in both the generation of mucous metaplasia in chronic bronchitis and the destruction of lung tissue in emphysema.
Neutrophil products induce mucus hypersecretion by both an acute secretagogue effect and by augmentation of the bronchial mucus producing apparatus.26,27 Mucin gene expression has been proposed as the principal factor governing the differentiation of epithelial cells into goblet cells.28 Neutrophil elastase (NE) and reactive oxygen species independently increase epithelial mucin mRNA and protein expression in vitro,29–31 possibly via ligand independent transactivation of the cell surface epidermal growth factor receptor (EGFR).29 In asthma the epithelial mucin, MUC5AC, and the EGFR are co-localised in airway epithelial goblet cells.32 As severe asthma is associated with both massively increased mucus production and neutrophilic airway inflammation,33–36 it has been hypothesised that neutrophil driven goblet cell metaplasia is a component of asthma airways remodelling. In COPD, MUC5AC is the principal secreted mucin of the epithelium37 and, like severe asthma and other hypersecretive diseases such as bronchiectasis and cystic fibrosis, neutrophilic airways inflammation is a key disease feature.13,33,34,38,39 Figure 1 demonstrates epithelial MUC5AC immunochemical staining in a bronchial biopsy specimen taken from a smoker with COPD, indicating its clear expression.

MUC5AC expression in the bronchial epithelium of a smoker with COPD. The photomicrograph shows immunohistochemical MUC5AC staining using an anti-MUC5AC antibody in a GMA embedded bronchial biopsy specimen.
The neutrophil’s standing as a potential key cell in emphysema has been assured since inherited α1-antitrypsin deficiency was first linked with the disease. The theory emerged that the neutrophil was the perpetrator of a protease/antiprotease imbalance in the lung.25 To support this, animal model studies have shown that administration of purified NE produces emphysema,40 while deficiency of endogenous NE affords protection against emphysema following exposure to cigarette smoke.41 Human studies have demonstrated NE in emphysematous tissue,42 increased products of elastase activity in urine and plasma from patients with COPD,43 and a correlation between emphysema severity and elastase levels in peripheral blood neutrophils.44 In vitro, peripheral blood neutrophils from subjects with emphysema digest more extracellular connective tissue protein than those taken from controls.45
The in vivo evidence linking neutrophils with COPD is abundant. Analyses of induced sputum14,15,46 and airway lavage fluid22,47,48 consistently demonstrate increased neutrophil counts and neutrophil derived enzyme levels in COPD, both when stable and during exacerbations.46,49–53 Within the airway wall, however, confirmation of neutrophilic airway inflammation has proved more elusive. Some investigations showing airway wall neutrophilia16,18,54 are countered by others that do not,17,19,22 an inconsistency that may reflect the short tissue lifespan of the infiltrating neutrophil. More recent studies have diversified their focus in an attempt to “capture” the elusive neutrophil. Bronchial biopsy specimens taken during a COPD exacerbation revealed higher neutrophil counts than those taken in stable disease.55 Analyses of airway smooth muscle have shown a relationship between neutrophil infiltration and both computed tomographic measurements of air trapping56 and severity of airflow obstruction.57 The investigators speculated that exposure to inflammatory mediators could affect the structure and contractility of airway smooth muscle, contributing to peripheral airways obstruction. This complements reports of a strong relationship between peripheral airways dysfunction in COPD and sputum neutrophil counts.58 Figure 2 shows immunochemically stained neutrophils infiltrating the bronchial submucosa and epithelium in a bronchial biopsy specimen, consistent with this hypothesis.

Neutrophils infiltrating bronchial submucosa and epithelium in a smoker with COPD. The photomicrograph shows immunohistochemical cytoplasmic staining using an anti-myeloperoxidase antibody in a GMA embedded bronchial biopsy specimen.
Macrophages
Macrophages account for the majority of inflammatory cells recovered by airway lavage, regardless of whether or not the subjects are non-smokers, healthy smokers or smokers with airways disease.48,59 Compounds released by macrophages include reactive oxygen species, chemotactic factors, inflammatory cytokines, smooth muscle constrictors, mucus gland activators, and extracellular matrix proteins. Also included is an array of matrix metalloprotease enzymes (MMPs). These can, when combined, degrade a similar spectrum of proteins to neutrophil enzymes25 and they are also believed to facilitate leucocyte migration and infiltration into injured tissues.60 Although macrophages do not transcribe the NE gene, their ability to internalise the enzyme has led to the proposal that macrophage released NE can add further to the proteolytic potential of the cell.61
Although macrophages have the theoretical potential to induce mucus hypersecretion via products with secretagogue activity such as leukotriene B4 (LTB4) and interleukin 1 (IL-1), most studies have focused on their possible role in emphysema, particularly via MMP production. In animal models administration of aerosolised alveolar macrophages produces emphysema,40 as does overexpression of MMP-1.62 Use of a broad spectrum MMP inhibitor attenuates both the initial inflammatory response and the severity of emphysema in guinea pigs exposed to cigarette smoke.63 In vitro, cultured alveolar macrophages taken from subjects with COPD express increased amounts of both MMP-1 and MMP-961 while, in vivo, immunoreactivity for MMP-2 and MMP -9 is demonstrably increased in COPD.64
MMP-12, in particular, has been a focus of increasing attention in emphysema. In mice, deficiency of MMP-12 appears to be protective against cigarette smoke induced lung destruction.65 In vivo, airway and alveolar macrophage MMP-12 expression in COPD subjects is enhanced compared with healthy controls.66,67 Recent evidence, moreover, suggests that the role of MMP-12 may go beyond matrix degradation. In mouse models its presence is required for cigarette smoke induced neutrophil influx into the lung,68,69 possibly via release of the pro-inflammatory cytokine tumour necrosis factor α (TNF-α) with subsequent upregulation of vascular adhesion molecule VCAM-1.70 This supports the view that neutrophils and macrophages are co-dependent on one another to produce an elastolytic inflammatory response in airways exposed to cigarette smoke.70
Studies of emphysematous lung tissue from human subjects have shown a direct relationship between alveolar macrophage density in the parenchyma and the severity of lung destruction.71 Moreover, numerous investigations have found macrophage numbers to be increased in the bronchial submucosa,14,16,17,19,72 bronchial glands,18 and small airways epithelium73 of subjects with COPD. Macrophage counts in induced sputum or bronchoalveolar lavage (BAL) fluid in COPD vary depending on whether relative or absolute cell counts are used. Relative macrophage counts are often reduced,14,15,74 a reflection, most likely, of increased neutrophil percentages. Absolute counts, on the other hand, are increased both in sputum46 and BAL fluid.48,59,75
T lymphocytes
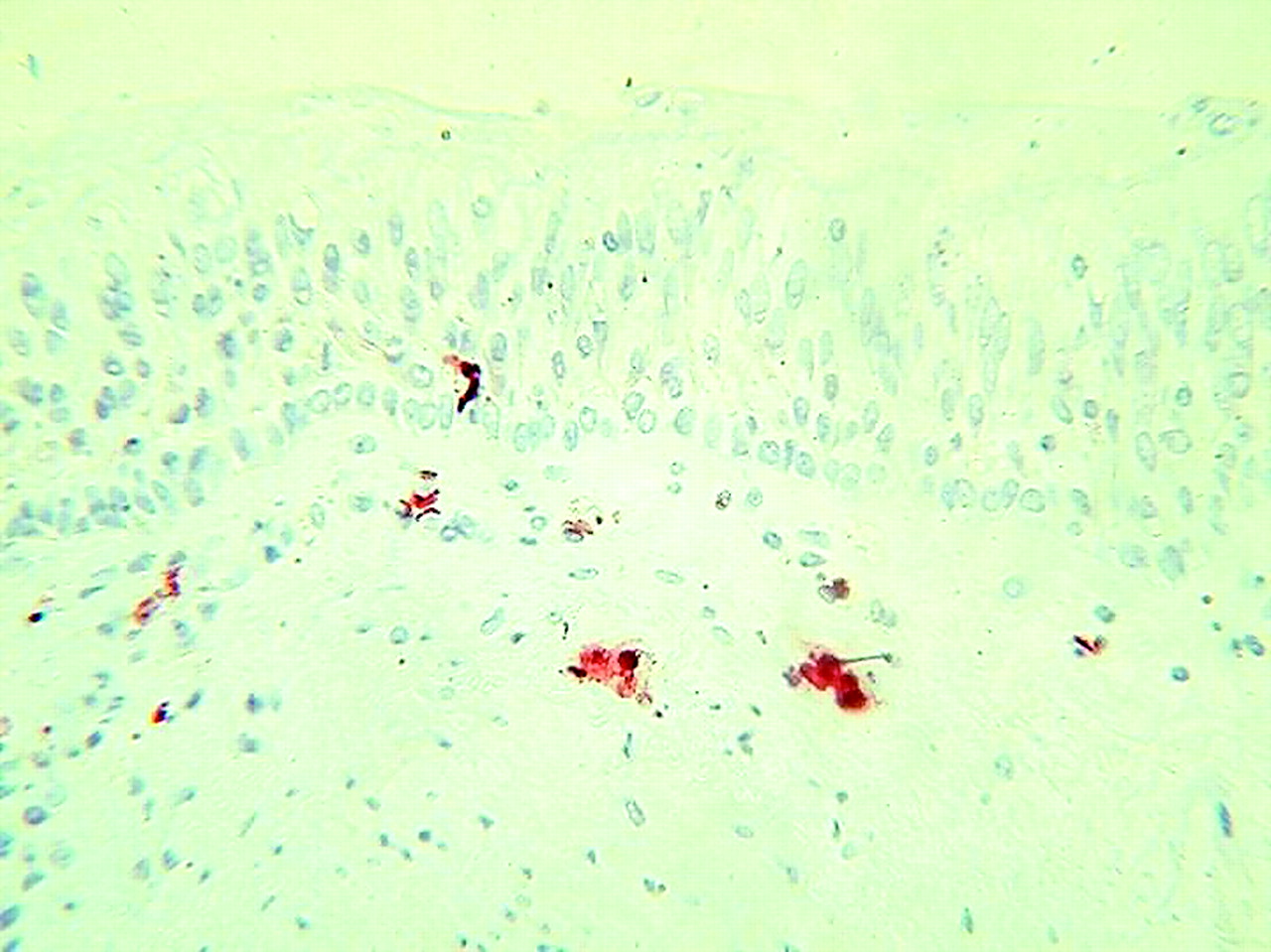
In asthma the CD4+ T cell is the proposed orchestrator of a Th2 type immune response in the airways.76 In COPD the CD8+ cell is the accepted crucial lymphocyte subtype. An increase in total T cells (CD3+ cells), which includes both CD4+ and CD8+ cells, occurs in the alveolar walls in emphysema,10,21 but CD8+ cells are predominant.21 Increased CD8+ cell numbers are also found in both the large19 and small20,73 airway walls in COPD and in peripheral airway smooth muscle.57 Figure 3 shows CD8+ cells infiltrating the bronchial submucosa and epithelium in a bronchial biopsy specimen.

{kind=link}
{kind=link}
{kind=link}
CD8+ cells infiltrating both submucosa and epithelium in a smoker with COPD. The photomicrograph shows immunohistochemical membrane staining using an anti-CD8 antibody in a GMA embedded bronchial biopsy specimen.
Speculation has surrounded the role of CD8+ cells in emphysema. A key function of the CD8+ cell is to combat viruses either by cytolysis of infected cells or induction of apoptosis.77,78 Collateral tissue damage is a possible consequence; in respiratory syncytial virus (RSV) infected mice, for example, excessive CD8+ cell activation results in potentially lethal pulmonary damage.79 In smokers infected with human immunodeficiency virus (HIV), high CD8+ cell numbers in BAL fluid have been associated with an accelerated onset of emphysema.80
If CD8+ cells destroy lung parenchyma, the mechanism is uncertain. They have, in theory, the potential to damage the lung interstitium directly via release of lytic substances such as perforin and granzyme.81 It was observed recently that CD8+ cells recovered from the sputum of COPD patients display higher levels of perforin expression and increased cytotoxic activity than CD8+ cells taken from control subjects.82 Another possibility is that CD8+ cells induce structural cell apoptosis.21 Alveolar cell turnover does appear to increase in emphysema as increased numbers of alveolar epithelial and endothelial cells undergo both proliferation and apoptosis.83–85 Moreover, an association has been observed between apoptosing cell numbers and CD8+ T cell numbers in the alveolar walls.21 The possibility remains, however, that such associations are a secondary phenomenon. Given their potential suppressor function, the infiltrating CD8+ cells may actually serve to inhibit the inflammatory process. Further studies are needed to define the activity of infiltrating CD8+ cells in emphysema.
Prior activation of T cells by antigen presentation is generally required before they can infiltrate non-lymphoid tissue such as the lung. Although the nature of the proposed antigen in COPD is unknown, two hypotheses have been put forward. Enelow and colleagues demonstrated that recognition of a lung “autoantigen” by T cells can produce lung injury in the absence of a viral stimulus, and this damage was mediated or amplified by non-antigen specific inflammatory cells such as macrophages.86 It was therefore hypothesised that repetitive injury to the lung, as a result of chronic smoking and inflammation, results in structural alteration of self-antigens allowing them to be recognised by T cells.21 Alternatively, a persistent intracellular pathogen may provide a foreign antigenic stimulus. One such candidate is adenovirus, as suggested by Retamales and colleagues87 who found a 41-fold increase in alveolar epithelial cells expressing the adenoviral transactivating protein E1A in subjects with severe emphysema compared with healthy smokers.
A number of molecules have been proposed to mediate pulmonary T cell infiltration in COPD. RANTES (regulated upon activation, normal T cell expressed and secreted), a potent chemoattractant that is overexpressed in exacerbations of chronic bronchitis,88 is believed to act synergistically with CD8+ cytolytic cells to enhance apoptosis of virally infected cells.89,90 It was therefore postulated that, when CD8+ cells predominate, recurrent viral infections with resulting increased RANTES levels may promote CD8+ cell mediated tissue damage.88 Th1 type cycle of inflammation has also been suggested; in COPD airways infiltrating CD8+ cells express more of the chemokine receptor CXCR3, its ligand CXCL10, and the inflammatory cytokine interferon γ (IFN-γ). This has led to the suggestion that an inflammatory insult induces increased airway expression of CXCL10 and IFN-γ, that this recruits CXCR3 bearing CD8+ cells, and that these—via expression of IFN-γ—could induce further CXCL10 expression thus perpetuating a cycle of CD8+ inflammation.91
The function of CD4+ cells in COPD is unknown. CD4+ cells, in addition to CD8+ cells, are increased in the small airway walls of smokers with severe COPD.73 They have the potential to contribute to the inflammatory process via production of a variety of pro-inflammatory cytokines including TNF-α and granulocyte-macrophage colony-stimulating factor (GM-CSF), the Th1 agents IFN-γ and IL-2, and the Th2 agents IL-4, IL-5, and IL-6. It has also been hypothesised that their actions as T helper cells, priming CD8+ cytotoxic responses, maintaining their memory and ensuring their survival, may be as important to the development of emphysema as the CD8+ cells themselves.21
Natural killer (NK) T lymphocytes
NK cells are a distinct population of specialised cytotoxic lymphocytes that target transformed or virus infected cells.92,93 Although the function of NK cells in COPD has not been widely studied, one investigation suggested that they are found in increased numbers in the large airway submucosa of smokers with COPD, with the hypothesis that excessive NK cell recruitment in COPD occurs due to repeated viral or bacterial infections.16 However, a study measuring lymphocyte subpopulations in the alveolar walls of non-smokers, healthy smokers, and smokers with emphysema found no increase in NK cell numbers in subjects with emphysema.21
Eosinophils and mast cells
Although eosinophils and mast cells are important effector cells in asthma, neither has been ascribed a prominent role in COPD. In exacerbations of COPD airway wall eosinophilia has been observed at a level similar to that found in asthma,88,94 with an associated increase in RANTES expression by airway subepithelial and epithelial cells.88 In stable COPD, increased eosinophil counts have been found in sputum,74 BAL fluid,48 and in the airway wall.22,54,95 Increased levels of eosinophilic cationic protein (ECP) in BAL fluid96 and induced sputum46,97,98 have also been observed. However, some studies have failed to confirm that any airway eosinophilia occurs in COPD,15,17–19,22,99 while others have suggested that the eosinophils, even if they are present, are not activated.14,22 The mechanism underlying eosinophilic infiltration seems to differ from asthma in that the key chemokines IL-4, IL-5 and eotaxin do not appear to be overexpressed in COPD airways.54 It has been hypothesised that any influx of these cells is merely a by-product of smoking induced inflammation. A possible mediator is the chemokine IL-8. Although normally associated with neutrophil chemotaxis, IL-8 exerts a chemotactic effect on primed eosinophils100,101 and correlates with BAL fluid levels of ECP in chronic bronchitis.52
Some reports have shown that increased numbers of eosinophils in sputum74,102,103 and BAL fluid104 taken from COPD subjects are predictive of a clinical response to steroid treatment. These results might suggest that eosinophils play a role in a clinical subset of COPD that has inflammatory features in common with that of asthma. However, the two diseases remain, in general, phenotypically dissimilar as even subjects with asthma who have fixed airflow obstruction have significantly higher percentages of eosinophils and lower percentages of neutrophils in both sputum and BAL fluid than COPD patients with similar disease severity.105
The ability of mast cells to release an array of mediators has invited speculation regarding their possible role in COPD inflammation. This could include neutrophil recruitment via the release of chemotactic factors, tissue injury by the actions of the secreted enzymes tryptase, chymase and elastase, and mucus hypersecretion via the potent secretagogue action of mast cell chymase.106 Some studies of COPD airways have revealed increased mast cell numbers in the airway wall.107,108 In addition, one study which found that mast cell degranulation in the bronchial gland layer appeared more marked in subjects with chronic bronchitis than in healthy controls suggested that increased mast cell activity may also be present.108 These findings are countered, however, by reports displaying no evidence of mast cell abundance in either the airways or parenchyma in COPD subjects.16–18,73,94,99
OBSTACLES TO RESEARCH INTO COPD INFLAMMATION
A detailed profile of disease phenotype specific inflammation has yet to emerge in COPD. There are many reasons for this, but chief among them may be the very nature of the condition itself. COPD is a heterogeneous disease, so the pathological significance of inflammatory cell measurements is often difficult to divine. The clinical severity varies considerably, asymptomatic disease is common,109,110 and interobserver variability exists in the detection of physical signs allowing mild cases to go undetected.111,112 Moreover, none of the common clinical tests give a complete picture of the disease. Spirometric tests are inadequate for the detection of early COPD,113,114 airflow obstruction severity is a poor predictor of symptom severity,115 and impairment of carbon monoxide gas transfer is not specific to emphysema and may occur by separate mechanisms in smokers.116–119
Linkage of lung function readings to the underlying pathology is hampered by the fact that both peripheral airway remodelling4,120,121 and emphysema6,7,122 result in airflow obstruction while emphysema is commonly found in smokers who have not developed airflow obstruction at all.113,114 Thus, characterisation of research subjects based only on clinical impression and lung function—as was the case with many studies14,20,95—fails to provide a clear insight into the underlying pathology. Neutrophil studies provide a prime example of the importance of distinguishing COPD phenotypes. Sputum neutrophil counts correlate with the severity of airflow obstruction in COPD,15,19 and smokers with severe emphysema have neutrophilic inflammation in the alveolar walls and air spaces.87 However, in mild emphysema there is neither BAL49 nor alveolar neutrophilia,87 even though BAL fluid levels of neutrophil degranulation products and the neutrophil chemokine IL-8 are increased.49,123 Mild emphysema would often go undetected on lung function testing so, without the use of CT scanning, such patients would be classified as healthy smokers.
A further obstacle is that the most practical investigative techniques have natural limitations. The predominant pathology is found in the not easily accessible peripheral airways and lung parenchyma. To what extent proximal airway biopsies represent events in the distal lung is uncertain. Although BAL and induced sputum sample more distally, the correlation between luminal and tissue inflammation is also unknown. Moreover, the results depend on the protocol in use. Inflammatory cell numbers vary depending on the lung region sampled, with neutrophils more numerous in the proximal bronchial tree and macrophages predominant more distally. Digital subtraction angiography has shown that BLF (fluid aspirated back from the initially instilled aliquots) samples proximal airways, whereas successive aliquots (BAL fluid) may reflect distal airway and alveolar events.124 Thus, in healthy smokers neutrophil percentages decline in successive BAL fluid aliquots.125 Sputum induction, similarly, is prone to sampling effects. In subjects with COPD, induced sputum contains higher percentages of neutrophils and lower percentages of macrophages than BAL fluid, most probably because the more proximal airways are sampled.14,126 However, increasing the duration of the induction results in a progressive reversal of these proportions, possibly due to increasingly peripheral airways sampling.127 Standardisation of induction duration is therefore important if reliable comparisons are to be made between different centres.
Prominent studies of airway inflammation in COPD have had conflicting results. Some investigations which showed CD8+ T lymphocyte infiltration of the airways19,20 have not been confirmed by others.14,16 Macrophage infiltration has similarly been both confirmed17,59 and refuted,20,95 while studies measuring neutrophils, eosinophils, and mast cells in airway wall and sputum have also produced inconsistent results.
The potentially crucial impact of infection and exacerbations on airways inflammation and disease severity remains unquantified. Frequent exacerbations of COPD are associated with a faster decline in forced expiratory volume in 1 second (FEV1) over time.128 Exacerbations are also associated with raised sputum counts of lymphocytes, neutrophils, and eosinophils.129 In the airway wall, similarly, both neutrophilia and eosinophilia are observed, as is overexpression of related chemokines IL-8, CXCL5 and RANTES.55,88,94 Bacterial infections, specifically, have been linked to increases in both neutrophil activation markers and associated inflammatory mediators including myeloperoxidase (MPO), NE, LTB4 and IL-8.130,131 Even in the absence of acute infection, airway bacterial colonisation is well recognised in COPD.132 Such colonisation has been correlated with airway neutrophil activation,133 sputum levels of the pro-inflammatory cytokines IL-8, LTB4 and TNF-α,134,135 airflow obstruction severity,136 and a more rapid decline in FEV1 over time in smokers.137 It has been hypothesised that chronic lower airway bacterial colonisation may accelerate the development of airflow limitation via both increased airway inflammation in stable disease and by predisposing to more frequent exacerbations.133,137,138 Although most studies relating inflammation to clinical disease severity have excluded acute infection from their subjects on clinical grounds, airway colonisation has not been ruled out to the same extent.
The effects of current smoking remain uncertain. Most investigators have preferred to record the pack year history instead. Rennard and colleagues showed that short term smoking reduction is associated with a reduction in airway inflammation in heavy smokers.139 Willemse and colleagues found a positive association between current smoking and macrophage numbers in both sputum and bronchial biopsy specimens.140 Other investigators have shown, however, that current and ex-smokers with COPD have similar degrees of inflammation in bronchial biopsy141 and induced sputum142 samples. Thus, although intuitively one would expect the ongoing insult to the airways to be relevant, the jury remains out as to whether differences in current smoking contribute to the variability of results between different studies.
Thus, to date, diverse methods and, often, diverse results, have contributed to uncertainty surrounding the inflammatory profile of COPD. If future anti-inflammatory strategies are to be tested, then measurements of inflammation before and after treatment in bronchoscopic or sputum samples may be necessary. More clarity is needed as to the strength of the relationships between proximal airway inflammation, distal airway inflammation, and lung pathology.
CONCLUSION
That airway inflammation is central to the pathogenesis of COPD appears beyond doubt. However, studies to date have varied widely in terms of the depth of disease characterisation, their allowance for confounding factors such as current smoking and infection, and the investigative methodologies employed. Future studies need to clarify the relationship between each specific inflammatory cell type and each of the remodelling and destructive processes found in COPD airways. Further research combining well validated techniques—specifically, comparison of preoperative endobronchial biopsy samples with examination of resected lung specimens and careful patient phenotyping—may help.
REFERENCES
Footnotes
-
Funding: none.
-
Competing interests: none.