Article Text

Abstract
Lymphocytic interstitial pneumonia (LIP) is a rare form of interstitial lung disease. A few case reports have described an association with common variable immunodeficiency (CVID). Corticosteroids are usually used to treat symptomatic patients but their efficacy has never been studied in a controlled trial. We describe a patient with LIP and CVID who was treated monthly with intravenous immunoglobulins (IVIG) without steroids. The patient improved dramatically. We believe that, in selected cases of LIP and immunodeficiency, IVIG given monthly should be considered as the only treatment without adding steroids.
- BOOP, bronchiolitis obliterans organising pneumonia
- CVID, common variable immunodeficiency
- IVIG, intravenous immunoglobulins
- LIP, lymphocytic interstitial pneumonia
- interstitial lung disease
- lymphocytic interstitial pneumonia
- common variable immunodeficiency
- immunoglobulins
- corticosteroids
Statistics from Altmetric.com
- BOOP, bronchiolitis obliterans organising pneumonia
- CVID, common variable immunodeficiency
- IVIG, intravenous immunoglobulins
- LIP, lymphocytic interstitial pneumonia
- interstitial lung disease
- lymphocytic interstitial pneumonia
- common variable immunodeficiency
- immunoglobulins
- corticosteroids
Lymphocytic interstitial pneumonia (LIP) is by definition a reactive lymphoid infiltrate which occurs most commonly in the alveolar septa but occasionally appears along bronchi and vessels.1 It may occur in association with a number of conditions including HIV in children, primary immunodeficiency, Sjögren’s syndrome, myasthenia gravis, and dysproteinaemic states including hyper- and hypogammaglobulinaemia.2 Hypogammaglobulinaemia occurs in about 10% of adults with this condition and the association of LIP with common variable immunodeficiency (CVID) has been described.3
The optimal treatment for LIP is not well established. Most patients are treated with prolonged courses of corticosteroids. We describe a patient who was not treated with steroids.
CASE REPORT
A 66 year old woman was admitted to the internal medicine ward for evaluation of worsening dyspnoea, fever, and productive cough. An antibiotic trial with amoxycillin+clavulanic acid and roxithromycin given by the family physician did not help. One year before admission the patient developed recurrent episodes of sinusitis, pneumonia, and bronchitis. A work-up done by the family physician revealed CVID with low levels of IgA and IgG2.
On admission the physical examination was normal with no clinical signs of Sjögren’s syndrome or other autoimmune diseases. The saturation was 94% and the lungs were clear. Complete blood count, chemistry panel, liver function tests, and urine analysis were normal. Arterial blood gas analysis on room air revealed Sao2 94%, Pao2 9.47 kPa (71 mm Hg), Paco2 4.27 kPa (32 mm Hg), HCO3 22.3 mm Hg, and pH 7.46. Antinuclear antibody, C-ANCA, P-ANCA, and rheumatoid factor were all negative. HIV and EBV serological tests were negative. The level of IgA was less than 42 mg/dl (normal 90–450) and the level of IgG2 was 86 mg/dl (normal 139–554). The levels of other immunoglobulins were within normal limits.
Spirometric parameters were normal and the chest radiograph showed interstitial markings. A computed tomographic (CT) scan of the chest showed acinary pulmonary nodules and ground glass opacities in both lungs (fig 1). Sputum cultures and throat swabs were negative. No acid-fast bacilli were seen. Bone marrow aspiration and biopsy were normal. Flexible bronchoscopy did not reveal endobronchial lesions. The bronchoalveolar lavage (BAL) fluid showed no infection or malignancy; a cell count was not performed. Transbronchial biopsy specimens displayed aggregates of small lymphocytes on the lung parenchyma. A specific diagnosis was not possible.
{kind=link}
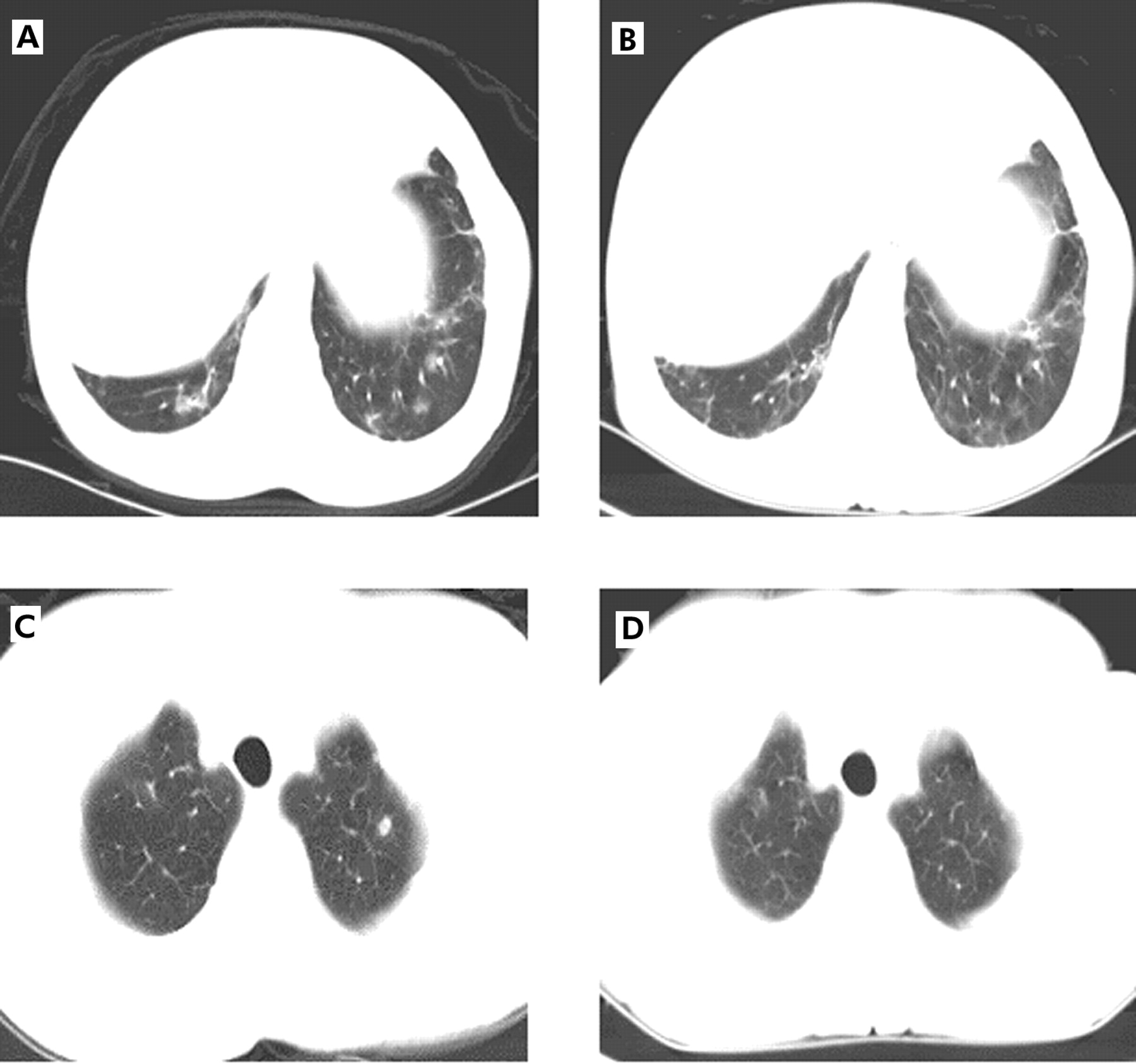
(A) CT scan (6.5 mm collimation) at the level of the diaphragm showing peribronchial thickening and ground glass opacities. (B) CT scan at the same level 5 months later showing partial resolution of the findings. (C) CT scan at the level of the trachea showing two ill defined nodules in the left apex. The largest nodule has central lucency which may represent a cavitation or a bronchial lumen surrounded by the nodule. There are focal ground glass opacities in the right apex. (D) CT scan at the same level 5 months later showing partial improvement. Two left apical nodules have resolved but there are several tiny ill defined nodules bilaterally. Focal ground glass opacities in the right apex are unchanged.
An open lung biopsy was performed and showed multifocal interstitial lymphoid infiltrates spreading into the alveolar septa and surrounding airways and vessels. The infiltrates were composed of small lymphocytes admixed with plasma cells. The lymphocytes were a mixture of polyclonal B cells (CD20 positive, mainly in nodules) and T cells (CD3 positive, mainly in pulmonary interstitium). Foci of bronchiolitis obliterans organising pneumonia (BOOP) were seen. These findings were consistent with LIP and associated BOOP. Cell rearrangement excluded monoclonality. The mixture of B and T cells combined with the cell rearrangement excluded the diagnosis of lymphoma.
Treatment with corticosteroids was considered but we could not find evidence to support this approach. The risk of severe infections and side effects of steroids made this treatment unfavourable. Treatment was therefore started with IVIG 0.5 mg/kg monthly. Seventeen months after the diagnosis the patient improved dramatically. She had only one episode of pneumonia compared with five episodes during the previous year. The chronic cough and dyspnoea resolved and Pao2 rose from 9.47 kPa (71 mm Hg) before the diagnosis to 11.87 kPa (89 mm Hg). The A–a gradient fell from 26 mm Hg before the diagnosis to a normal level (7 mm Hg). Pulmonary function tests remained normal and the CT scan showed partial resolution of the findings (fig 1A–D).
DISCUSSION
We describe a patient suffering from CVID and LIP. Monoclonality in the cell populations, which would support a diagnosis of lymphoma, was excluded. The patient was treated with IVIG without steroids.
The optimal treatment for LIP is not well established and there are no controlled trials. However, most patients are treated with prolonged courses of corticosteroids which may control the process or induce remission. Popa et al4 described two patients with LIP associated with CVID who were treated with IVIG, both of whom died after severe infections. They also described patients with other forms of interstitial lung disease who improved after treatment with IVIG.4 De Gracia et al5 used IVIG to treat 24 patients diagnosed with CVID; patients suffering from chronic lung disease showed improvement with this treatment.
Our patient showed significant improvement following treatment with IVIG. The mechanism behind the effect of IVIG is not known. We speculate that recurrent infections cause a chronic inflammatory process which continues as a result of immunoglobulin deficiency. There is a possibility that replacement therapy with immunoglobulins might prevent infections, decrease the exposure to antigens, and stop the inflammatory process. Another possibility is immunomodulation by changing the CD4/CD8 ratio in the lung interstitium.6
We believe that, in patients with LIP and immunodeficiency, IVIG given monthly without steroids may be a preferred treatment option. Steroids have severe side effects and might expose these patients to infections. They should only be considered when the patient shows clinical and radiological progression of the interstitial lung disease.
Footnotes
-
Competing interests: none.