Article Text

Abstract
Background: Chronic bird fancier’s lung (BFL) has often been misdiagnosed as one of the idiopathic interstitial pneumonias (IIPs).
Methods: To define the clinical and pathological characteristics of chronic BFL, 26 patients with chronic BFL from whom a surgical lung biopsy specimen was taken between October 1992 and June 2001 were evaluated. The histopathological characteristics of the surgical lung biopsy specimens were examined and correlations between the histopathology and clinical characteristics were analysed. The quality of chronic inflammatory and fibrotic changes was expressed according to the 2002 ATS/ERS consensus classification of IIPs.
Results: Two patients were diagnosed as having bronchiolitis obliterans organising pneumonia (BOOP)-like lesions, five as having cellular non-specific interstitial pneumonia (NSIP)-like lesions, and eight as having fibrotic NSIP-like lesions. The other 11 patients were considered to have usual interstitial pneumonia (UIP)-like lesions because of the temporal heterogeneous appearances of the fibrotic changes. However, fibrosis in these patients had developed in centrilobular as well as perilobular areas, suggestive of hypersensitivity pneumonitis. Nineteen patients (73.1%) had multinucleated giant cells, often with cholesterol clefts, while only five patients (19.2%) had granulomas. Patients with BOOP-like or cellular NSIP-like lesions tended to have recurrent acute episodes, whereas patients with UIP-like lesions had an insidious onset. Patients with BOOP-like or cellular NSIP-like lesions had a more favourable outcome than those with fibrotic NSIP-like and UIP-like lesions.
Conclusions: The qualities of chronic inflammatory and fibrotic lesions vary significantly among patients with chronic BFL but correlate with clinical features and prognosis.
- BFL, bird fancier’s lung
- BOOP, bronchiolitis obliterans organising pneumonia
- HP, hypersensitivity pneumonitis
- IIP, idiopathic interstitial pneumonia
- NSIP, non-specific interstitial pneumonia
- UIP, usual interstitial pneumonia
- bird fancier’s lung
- hypersensitivity pneumonitis
- usual interstitial pneumonia
- non-specific interstitial pneumonia
- bronchiolitis obliterans organising pneumonia
Statistics from Altmetric.com
- BFL, bird fancier’s lung
- BOOP, bronchiolitis obliterans organising pneumonia
- HP, hypersensitivity pneumonitis
- IIP, idiopathic interstitial pneumonia
- NSIP, non-specific interstitial pneumonia
- UIP, usual interstitial pneumonia
- bird fancier’s lung
- hypersensitivity pneumonitis
- usual interstitial pneumonia
- non-specific interstitial pneumonia
- bronchiolitis obliterans organising pneumonia
Hypersensitivity pneumonitis (HP) comprises a group of allergic lung diseases caused by inhalation of various antigens contained in organic dusts. Bird fancier’s lung (BFL) is a form of HP resulting from airborne exposure to avian antigens.1,2 In most cases of HP histological confirmation is unnecessary.3 Histopathologically, HP is characterised by features of a granulomatous interstitial pneumonia—namely, chronic inflammatory infiltrates along the small airways (cellular bronchiolitis); diffuse interstitial infiltrates of chronic inflammatory cells; and scattered, small, non-necrotising granulomas.3 This triad is found in most but not all cases.3–7 An epidemiological survey of chronic HP cases in Japan revealed that granulomas were usually not observed in BFL (16.7%); they were more frequent in summer type HP (44.4%) and were prevalent in isocyanate HP (60.0%).8 Previous studies of the pathology of HP focused almost entirely on farmer’s lung.5–7 The pathology of BFL was reported by Perez-Padilla et al.9 These studies were published before the concept of non-specific interstitial pneumonia (NSIP) was developed.10
An international classification of idiopathic interstitial pneumonias (IIPs) has been proposed by the American Thoracic Society (ATS) and the European Respiratory Society (ERS).11 Usual interstitial pneumonia (UIP) is the histopathological presentation that is associated with idiopathic pulmonary fibrosis (IPF).11,12 In the original report10 NSIP included interstitial pneumonia, either idiopathic or secondary, associated with disorders such as collagen vascular diseases or exposures. Further, two subgroups of NSIP were categorised histologically with either a cellular or a fibrotic NSIP pattern.11,13,14
Recent articles have reported that the histological features of chronic BFL can be UIP-like15 or NSIP-like.16 In this study we applied the histopathological description of the ATS/ERS international consensus classification of the IIPs to the chronic inflammatory and fibrotic lesions in chronic BFL, and related the histological changes to the clinical course and outcome of our patients clinically characterised in a previous study.17 Discussion of the histology of these cases was minimal in the previous study17 because the main focus of that study was on the clinical features of chronic BFL. In contrast, this paper focuses on the various histopathological patterns in chronic BFL.
METHODS
Subjects
The records and histopathological reports of 26 patients with chronic BFL (15 men) admitted to our hospital who had undergone surgical lung biopsy between October 1992 and June 2001 were examined. All the patients were referred to our hospital for clinical management. The diagnostic criteria for chronic BFL included (1) a history of avian contact, (2) antibodies and/or lymphocyte proliferation to avian antigens, (3) reproduction of symptoms of HP by an environmental provocation or laboratory controlled inhalation of avian antigens,18 either (4) evidence of pulmonary fibrosis with or without granulomas on histopathological analysis, or (5) honeycombing on computed tomographic (CT) scans, (6) progressive deterioration of a restrictive impairment on pulmonary function over 1 year, and (7) respiratory symptoms related to HP of 6 months duration.8,17,18 Honeycombing on HRCT scans is an alternative to the pulmonary fibrosis on histopathological analysis and was included in the diagnostic criteria as an alternative in the original study.8 Informed consent was obtained from all patients.
Histopathological evaluation
Lung tissues were obtained either by open lung biopsy or video-assisted thoracoscopic surgery, usually within 1 week of admission to hospital. Histological sections of biopsy materials were stained with haematoxylin-eosin and elastica van Gieson. The histological examinations were interpreted blindly by five pulmonary pathology specialists without knowledge of the patient’s clinical course. When the interpretations differed between the five pathologists, the final decision was reached by consensus.
The chronic inflammatory and fibrotic lesions were classified according to the ATS/ERS international consensus classification as UIP-like lesions, NSIP-like lesions, and bronchiolitis obliterans organising pneumonia (BOOP)-like lesions based on the quality of fibrotic changes including the loose and dense fibrosis, and the temporal appearance.10–14 The key histopathological difference between NSIP-like lesions and UIP-like lesions is an absence of temporal heterogeneity.10–14 Patients with NSIP were subdivided into two groups: cellular NSIP pattern and fibrotic NSIP pattern.11,13,14
Correlation of histopathology with clinical features and prognosis
In addition to the assessment of the histopathological pattern in the surgical lung biopsy specimens, we evaluated the relationship between these patterns and the clinical characteristics which included laboratory findings, pulmonary function tests, HRCT scans, bronchoalveolar lavage (BAL) findings, and response to treatment.
Inhalation challenge
Antigen inhalation provocation tests were conducted as previously reported.18 Briefly, patients inhaled 2 ml of pigeon dropping extract (PDE, 340 μg/ml) through a hand held nebuliser. The following monitoring parameters were used to determine the criteria for inhalation provocation challenge; patients who fulfilled three or more of the following were considered positive and those who fulfilled two were considered as probable positive: (1) increased radiological abnormalities; (2) an increase in the alveolar-arterial difference in oxygen tension (A-aDo2) of more than 10 mm Hg and/or a decrease in the carbon monoxide transfer factor (Tlco) of more than 20%; (3) a decrease in vital capacity (VC) of more than 15%; (4) an increase in peripheral leucocyte count of more than 30%; (5) an increase in C reactive protein (CRP) of more than 1.0 mg/dl; (6) an increase in body temperature of more than 1.0°C and/or the development of systemic manifestations including chills and general fatigue; and (7) the development of respiratory symptoms (cough and dyspnoea).
Immunological findings
Antibodies in sera and BAL fluids to PDE or budgerigar dropping extract (BDE) were measured by an enzyme linked immunosorbent assay (ELISA).19
Antigen induced lymphocyte proliferation was performed as previously reported.19 Peripheral or bronchoalveolar lymphocytes (2×105 cells) were cultured in quadruplicate with a 1:100 dilution of pigeon sera or budgerigar sera in 96-well flat bottom plates. The plates were incubated for 5 days at 37°C in a 5% CO2 incubator with 3H-thymidine for the last 16 hours of culture. The results were expressed as a stimulation index, which is the geometric mean counts per minute of stimulated cultures with pigeon or budgerigar sera divided by the geometric mean counts per minute of unstimulated cultures as control (medium only). Experiments showed that a positive stimulation index was ⩾2.0 (data not shown).
Therapeutic responses
The definition of a therapeutic response is as described in the ATS consensus report.12
Statistical analysis
Data are expressed as mean (SE). For statistical analysis, Stat View 5.0 and Statcel Statistical Software running on an Apple Macintosh personal computer were used. The results from the three groups were compared using the Kruskal-Wallis test. Post-hoc analysis between two variables was performed using the Scheffe test. Comparison between groups was performed using the χ2 or Fisher’s exact test for categorical variables. Cumulative survival probabilities were estimated using the Kaplan-Meier method. The log rank test was used to compare survival between the patients of each group. All statistical comparisons were two sided and were carried out at the 0.05 significance level.
RESULTS
Clinical features
All of the 26 patients fulfilled the abovementioned diagnostic criteria for chronic BFL. Chronic BFL was clinically divided into two subgroups: recurrent and insidious.17 The 10 patients with recurrent BFL showed reproduction of symptoms related to HP by environmental provocation at the beginning of the disease process, while the 16 patients with insidious BFL had a positive result following a laboratory controlled inhalation provocation test but not following environmental exposure. All of the patients with chronic BFL had pulmonary fibrosis on histopathological examination, and most of the patients classified as having a UIP-like pattern had honeycombing on the HRCT scan. Eleven of the 26 patients had been diagnosed as having IPF and one as having NSIP before the first visit to the hospital.
Histological classification
Of the 26 patients with chronic BFL (table 1), two (one man and one woman) were classified as having BOOP-like lesions (fig 1), five (two men and three women) were classified as having cellular NSIP-like lesions (fig 2A), eight (four men and four women) were considered to have fibrotic NSIP-like lesions (fig 3), and 11 (eight men and three women) were classified as having UIP-like lesions. The criteria for UIP-like lesions were heterogeneous appearance at low magnification with alternating areas of normal lung structure, scarce interstitial inflammation, dense fibrosis and honeycombing, and the presence of fibroblastic foci (fig 4A–C). Although only a small proportion of patients with UIP-like lesions had cellular bronchiolitis (table 2), fibrosis in these patients had developed in both centriacinar and perilobular regions, suggesting an HP-like pattern (fig 4B). However, one of the 11 patients with UIP-like lesions was indistinguishable from UIP/IPF except for the presence of multinucleated giant cells. Multinucleated giant cells with cholesterol clefts in their cytoplasm and lymphoid follicles were frequently observed (fig 2B, C and table 2). The findings of centriacinar fibrosis and multinucleated giant cells are inconsistent with IPF patients who have the histopathological pattern of UIP, suggesting an HP-like pattern. Granulomas were found in only five of the 26 cases (one with BOOP-like lesions, two with cellular NSIP-like lesions, and two with fibrotic NSIP-like lesions). No patient with chronic BFL had desquamative interstitial pneumonia (DIP)-like or diffuse alveolar damage (DAD)-like lesions.
Characteristics of study patients
Histopathological characteristics of surgical lung biopsy specimens in chronic bird fancier’s lung (BFL)

Bronchiolitis obliterans organising pneumonia (BOOP)-like lesions (patient 2 in table 1). Low magnification photograph of the surgical lung biopsy specimen showing patchy lung involvement by loose plugs of connective tissue within alveolar ducts and alveolar spaces. Stain: haematoxylin-eosin; original magnification ×18.

Cellular non-specific interstitial pneumonia (NSIP)-like lesions (patient 4 in table 1). (A) Low magnification photograph of the surgical lung biopsy specimen showing moderate to severe interstitial inflammation. (B) Higher magnification of left middle portion of (A) showing lymphoid follicle in bronchovascular bundle that was often seen in all groups. (C) Higher magnification of the right upper portion of (A) showing multinucleated giant cells with cholesterol clefts considered another finding suggestive of bird fancier’s lung. Stain: haematoxylin-eosin; original magnifications: A ×18, B ×50, C ×100.

Fibrotic non-specific interstitial pneumonia (NSIP)-like lesions (patient 11 in table 1). Low magnification photograph of the surgical lung biopsy specimen showing loose fibrosis with moderate infiltration of lymphoid cells, not only in subpleural areas but also in the centrilobular area. Stain: haematoxylin-eosin; original magnification ×18.

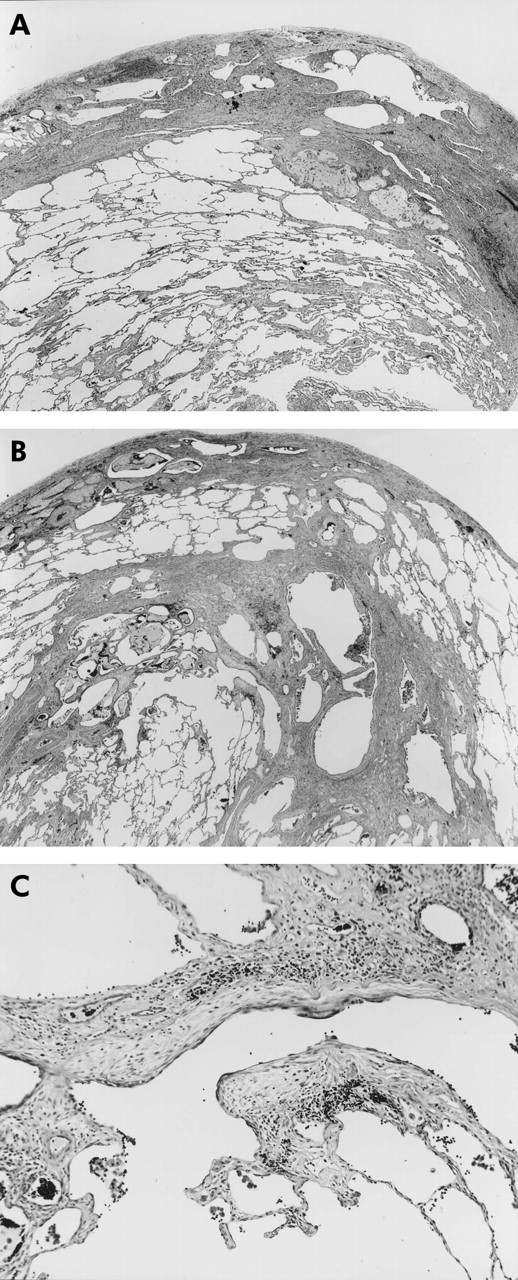
Usual interstitial pneumonia (UIP)-like lesions (patient 23 in table 1). (A) Low magnification photograph of the surgical lung biopsy specimen showing temporal heterogeneous appearance with alternating areas of normal lung structure, interstitial inflammation, dense fibrosis, and honeycomb changes which was indistinguishable from UIP/idiopathic pulmonary fibrosis (IPF). (B) Low magnification photograph of another portion of (A) showing not only subpleural fibrosis as seen in UIP but also distinct centrilobular fibrosis, which is inconsistent with idiopathic pulmonary fibrosis (IPF). (C) Higher magnification of the middle portion of (B) showing fibroblastic foci. Stain: haematoxylin-eosin, original magnifications: A ×22, B ×18, C ×190.
Clinical characteristics and prognosis according to histopathological group
The two patients with BOOP-like lesions and the five patients with cellular NSIP-like lesions were categorised as group A because neither of these lesions showed marked interstitial fibrotic lesions. Eight patients with fibrotic NSIP-like lesions were categorised as group B and 11 patients with UIP-like lesions were categorised as group C.
There were no significant differences in age, sex, smoking habit, antigen exposure periods, %VC, or %Tlco between the three groups (table 3). Patients in group A tended to have recurrent acute episodes including mild exertional dyspnoea, cough and low grade fever, whereas patients in group C experienced no acute episodes, suggesting that they presented with chronic disease of insidious onset. Specific antibodies were more frequently positive in group A patients than in group C patients. Antigen induced lymphocyte proliferation was frequently positive in all groups. The stimulation index for peripheral lymphocyte proliferation was higher in group A patients (8.1 (1.9)) than in group B patients (2.8 (0.3)) or group C patients (3.1 (0.4)). The stimulation index for antigen induced peripheral lymphocyte proliferation was 1.1 (0.1) in IPF patients (n = 21) and 1.3 (0.2) in asymptomatic bird owners (n = 20). Honeycombing on HRCT scans was more frequently observed in group C patients than in group A patients, although emphysema was not seen in never smoking patients in any of the groups. BAL lymphocytes were significantly lower in group C patients than in patients in groups A and B.
Clinical characteristics and histological pattern in patients with chronic bird fancier’s lung (BFL)
While all the 26 patients with chronic BFL had basically discontinued their antigen exposure, 23 were being treated with oral prednisolone depending on the severity of the initial signs and symptoms, with an initial daily dose of 30–40 mg for 2 weeks which was then tapered. One patient (no 12) refused treatment with prednisolone because of diabetes. The remaining two patients (nos 16 and 21) were not treated because they had no respiratory complaints and had come to the hospital because of abnormal chest shadows observed during a general examination. Patients in group A had a more favourable response to prednisolone treatment than those in groups B and C (table 3). Seven patients in group A, seven in group B, and nine in group C were followed for 83.7 (6.4), 64.4 (13.9), and 28.1 (5.6) months, respectively, after starting steroid treatment. Patients in group A had a significantly better survival than those in groups B and C (p = 0.016 and p = 0.015, respectively; fig 5). Ten patients (four in group B and six in group C) died 46.8 (10.8) and 30.5 (8.8) months, respectively, after surgical lung biopsy, despite steroid treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier survival curves for the three groups of patients: group A (bronchiolitis obliterans organising pneumonia (BOOP)-like or cellular non-specific interstitial pneumonia (NSIP)-like lesions, n = 7); group B (fibrotic NSIP-like lesions, n = 8); and group C (usual interstitial pneumonia (UIP)-like lesions, n = 11). Patients in group A had a significantly better survival than those in groups B and C (p = 0.016 and p = 0.015, respectively).
DISCUSSION
This clinicopathological correlation study was conducted in 26 patients with a diagnosis of chronic BFL based on clinical characteristics and a positive provocation test, the “gold standard” in diagnosing HP. The detailed clinical features other than the histopathology of these patients were described in a previous paper.17 We found that the histopathological patterns described in the 2002 ATS/ERS consensus classification of IIPs were significantly correlated with the clinical course of the disease, BAL findings and, in particular, with response to treatment and prognosis. Patients with BOOP-like or cellular NSIP-like lesions had a better outcome than those with fibrotic NSIP-like or UIP-like lesions.
Although granulomas are generally considered to be characteristic histological findings of HP, Reyes and colleagues6 reviewed the pathology of 60 cases of farmer’s lung and reported that interstitial alveolar infiltrates—consisting of plasma cells, lymphocytes, and occasionally eosinophils—were present in all cases while granulomas were found in only about 70%. Seal et al7 reported that the principal histopathological finding in six cases of chronic farmer’s lung was interstitial fibrosis without granulomas. Pathological studies of end stage BFL have shown that the alveoli are almost completely replaced by connective tissue and honeycombing, although no granulomas were found.20–22 In our study, granulomas were seen in only a small proportion of patients with chronic BFL.
The UIP pattern is the histopathological feature of IPF which is a clinicopathological disease entity. In our study, 11 patients with chronic BFL were found to have UIP-like lesions with a temporal heterogeneous appearance at low magnification and alternating areas of normal lung structure, scarce interstitial inflammation, fibrosis, and honeycomb changes as well as fibroblastic foci. These patients experienced no acute episodes and most of them had no specific antibodies and no lymphocytosis in the BAL fluid, although antigen induced lymphocyte proliferation tests and laboratory controlled inhalation provocation tests were positive. In fact, eight of these 11 patients had been diagnosed with IPF before visiting our hospital. However, the fibrotic changes were located in both centrilobular and perilobular areas, suggesting an HP-like pattern, whereas they are exclusively perilobular in IPF. Multinucleated giant cells with cholesterol clefts were also considered suggestive of BFL; these are rarely seen in IPF. Surgical lung biopsies on two separate occasions in a patient with chronic HP showed that the progression of chronic disease was associated with the disappearance of the centrilobular pattern leading to dense fibrosis.23 In addition, necroscopic specimens from patients with UIP-like lesions, who had fibrosis in both centrilobular and perilobular areas on specimens obtained by surgical lung biopsy, demonstrated widespread honeycombing which was indistinguishable from UIP.20 In fact, patients with UIP-like lesions were diagnosed as having UIP/IPF by a general pathologist, not by lung experts, before referral to our hospital. Current observations were the consensus results reviewed by five experienced Japanese lung pathologists; therefore, chronic insidious BFL is one disorder that can be difficult to differentiate from UIP/IPF. The international consensus statement by the ATS pointed out that a pattern of interstitial inflammation and fibrosis, sometimes indistinguishable from UIP, can occur in patients with chronic HP which is distinguished by clinical, serological, or radiographic manifestations.12 However, surgical lung biopsies should be conducted early enough to identify clearly the histopathological patterns of HP during the course of the disease.15
Idiopathic NSIP could be separated into cellular and fibrotic patterns because these histological patterns are associated with different clinical characteristics and prognosis.11,13,14 Patients with idiopathic cellular NSIP had a more favourable outcome than those with idiopathic fibrotic NSIP and IPF.11,13,14 In the current study, patients with BOOP-like or cellular NSIP-like lesions had a more favourable outcome than those with fibrotic NSIP-like lesions or UIP-like lesions as observed in idiopathic NSIP and IPF. Patients with chronic BFL who had UIP-like lesions were found to have a poor prognosis in a study from Mexico.24 BAL lymphocytosis was seen in patients with idiopathic NSIP and in chronic BFL patients with NSIP-like lesions, whereas the proportion of BAL lymphocytes in chronic BFL patients with UIP-like lesions (19.1 (2.9)%) was higher than in patients with UIP/IPF (7.2%) as reported by Nagai et al.13
Patients with BOOP-like or cellular NSIP-like lesions tended to have recurrent acute episodes, whereas patients with UIP-like lesions had an insidious onset. One patient with cellular NSIP-like lesions with insidious onset had a favourable response to steroid treatment and two patients with fibrotic NSIP-like lesions with recurrent episodes did not respond favourably to steroids. The primary determinant of the more favourable response to steroid treatment therefore seems to depend on the histological appearance rather than the clinical presentation.
Emphysema has been reported to occur frequently in patients with farmer’s lung, even in those who never smoked,25 and it is a more prevalent long term outcome of farmer’s lung than interstitial lung fibrosis.26 This is different from chronic BFL where lung fibrosis is the major outcome. In this study we applied the histopathological description of the ATS/ERS international consensus classification of IIPs to the chronic inflammatory and fibrotic lesions in chronic BFL, although emphysema has not been seen in patients with chronic BFL who never smoked.
The patients in our study had a wide range of histopathological findings. Jacobs et al27 reviewed consecutive cases of IIPs referred by pulmonologists in general practice and reported that UIP, BOOP, NSIP, and non-classifiable morphological patterns represented a spectrum of interstitial lung disease that may be caused by inhalation of organic dusts in the home or workplace as described with HP. They emphasised that all patients with IIPs must be suspected of having an HP until aggressive evaluation proves otherwise.15 Perez-Padilla et al9 compared the histopathology of chronic BFL, IPF, and UIP with bird exposure, although UIP with bird exposure in their population may represent patients with an advanced stage of BFL because they did not conduct antigen induced lymphocyte proliferation or laboratory controlled inhalation provocation tests. Our study shows that patients with chronic BFL can produce the whole range of histological features covered by the new consensus classification for idiopathic forms of diffuse interstitial lung disease. This suggests that these features are more a reflection of natural history at different stages of disease evolution or of differences in host responses than of fundamental differences in the pathology of different clinical diseases.
In conclusion, chronic BFL should be included in the list of differential diagnoses of interstitial lung disease, especially in patients with avian contact, even when granulomas are absent. A pathologist can suggest a diagnosis of chronic BFL on the basis of centriacinar fibrotic lesions at low power magnification. It is important to obtain a detailed clinical history and to perform adequate pathological diagnostic procedures for making a correct diagnosis of chronic BFL. Antigen induced lymphocyte proliferation and laboratory controlled inhalation tests are helpful to confirm the diagnosis and to exclude the possibility of IPF and idiopathic NSIP. The patterns of chronic inflammatory and fibrotic lesions are diverse among patients with chronic BFL but correlate with clinical features and prognosis.
Acknowledgments
The authors thank Dr Y Kobashi, Division of Pathology, Tenri Hospital, Dr T Takemura, Division of Pathology, Japan Red Cross Center, and Dr Y Kawabata, Division of Laboratory Medicine, Saitama Cardiovascular and Respiratory Center for discussions on histopathology; Dr T Shibata and Dr H Tanaka, Department of Bioinformatics, Tokyo Medical and Dental University for statistical analysis; and Dr V L Moore, Merck Research Laboratories (retired), Phillips, Wisconsin for the critical review of the manuscript.
REFERENCES
Footnotes
-
This work was supported by the Research Committee of the Japanese Ministry of Health and Welfare on Interstitial Pulmonary Disease, grant-in-aid for scientific research 12670552 from the Japanese Ministry of Education, the Research on Health Sciences focusing on Drug Innovation, and Charitable Trust of Okamoto Satoshi Fund for Fibrotic Lung Disorders.