Article Text

Abstract
The development of pulmonary hypertension is a poor prognostic sign in patients with chronic obstructive pulmonary disease (COPD), affecting both mortality and quality of life. Although pulmonary hypertension in COPD is traditionally viewed as a result of emphysematous destruction of the vascular bed and/or hypoxia, recent studies indicate that neither of these factors correlates very well with pulmonary artery pressures. New human and animal experimental data are beginning to show that pulmonary hypertension in this setting is probably a result of the direct effect of tobacco smoke on the intrapulmonary vessels with abnormal production of mediators that control vasoconstriction, vasodilatation, and vascular cell proliferation, ultimately leading to aberrant vascular remodelling and aberrant vascular physiology. These changes are in many ways similar to those seen in other forms of pulmonary hypertension and suggest that the treatments used for primary pulmonary hypertension may be beneficial in patients with COPD.
- eNOS, endothelial nitric oxide synthase
- ET-1, endothelin-1
- FEV1, forced expiratory volume in 1 second
- NO, nitric oxide
- Pao2, arterial oxygen tension
- VEGF, vascular endothelial growth factor
- chronic obstructive pulmonary disease
- pulmonary hypertension
- cigarette smoke
- vasoactive mediators
- emphysema
Statistics from Altmetric.com
- eNOS, endothelial nitric oxide synthase
- ET-1, endothelin-1
- FEV1, forced expiratory volume in 1 second
- NO, nitric oxide
- Pao2, arterial oxygen tension
- VEGF, vascular endothelial growth factor
- chronic obstructive pulmonary disease
- pulmonary hypertension
- cigarette smoke
- vasoactive mediators
- emphysema
Chronic obstructive pulmonary disease (COPD) has become the fourth leading cause of morbidity and mortality, accounting for half a million potential years of life lost and for more than $18 billion in direct costs in the USA and approximately $1.4 billion in the UK.1 In COPD patients, clinically diagnosed cor pulmonale has long been known to be an important negative prognostic sign, even after correction for forced expiratory volume in 1 second (FEV1).2 When pulmonary haemodynamics have been measured, an increase in the mean pulmonary arterial pressure at rest has also been shown to be an important prognostic variable,2 with an increase in the mean pressure of 10 mm Hg associated with a greater than fourfold increase in mortality.3
The actual incidence of pulmonary hypertension in COPD is not known, although early studies estimated that 6% of patients with COPD would develop cor pulmonale each year. A more recent study examined patients with COPD with FEV1 ranging from <35% predicted (28.2% of subjects) to >50% predicted (26.5% of subjects), none of whom had pulmonary hypertension at rest, over an interval of approximately 6 years.4 At the second assessment it was found that the mean pulmonary arterial pressure had increased slowly (0.28 mm Hg per year) with 25% of all patients now having a resting pulmonary arterial pressure of >20 mm Hg, and that this was more common in subjects who had pulmonary hypertension on exercise at the first assessment. This rate of progression was similar to the mean rate of increase of 0.6 mm Hg found in an earlier study.5 Extrapolation of these data suggests that a significant number of patients with COPD will develop pulmonary hypertension over the course of their disease and will have increased morbidity and mortality as a result.
It is important to note that the increases in pulmonary arterial pressure observed in patients with COPD generally tend to be modest. Based on analysis of data derived from a population of patients with idiopathic (“primary”) pulmonary arterial hypertension, the cardiopulmonary haemodynamic changes in COPD would not, in and of themselves, be expected to be associated with a strong effect on mortality. Thus, the fact that there is a distinct increase in mortality in patients with COPD and pulmonary hypertension suggests that a complicated set of interactions is occurring in the lung.
In patients with idiopathic pulmonary hypertension and pulmonary hypertension related to secondary causes such as connective tissue diseases, the hypertension has well recognised effects on functional capacity, exercise intolerance, and health related quality of life, quite apart from its effects on mortality. While abnormal ventilatory function and inadequate ventilatory reserve are common important factors limiting peak exercise capacity in patients with COPD, the same mechanisms that affect overall quality of life in patients with idiopathic pulmonary hypertension probably also apply to patients with COPD and pulmonary hypertension.
This paper is not meant to be an all encompassing review but, rather, a short reprise of the various theories put forward to explain the aetiology of pulmonary vascular dysfunction and the development of pulmonary hypertension in cigarette smokers. Most information about pulmonary hypertension in COPD has been derived from human studies and relatively few experimental animal models have examined the effects of smoke exposure on the pulmonary vasculature. However, it has been shown that chronic smoke exposure in outbred Hartley strain guinea pigs is associated with an increase in pulmonary arterial pressure, and this occurs only in a subset of the population,6 similar to the situation in humans. We consider experimental studies in greater detail below.
STRUCTURAL CHANGES IN THE PULMONARY VASCULATURE IN CIGARETTE SMOKERS
The structure of the muscular pulmonary arterial compartment in smokers has been examined extensively at necropsy and in surgical resection specimens. However, it is often difficult to use these reports to deduce pathophysiological mechanisms. While many studies examine pathological and/or physiological changes in subjects with COPD, others use the presence or absence of emphysema as a discriminator, while still others focus on the differences between tobacco smokers versus ex-smokers or non-smokers.
Studies of the vasculature of patients with moderate to severe COPD have consistently identified changes in the intima,7,8,9,10 with focal fibroelastic thickening and an increase in longitudinal muscle.9 The increased thickness of the intima has been shown to be caused by smooth muscle cell proliferation and increased elastin in addition to collagen deposition.10
The overall wall thickness (including intima and muscular media) of vessels of 100–200 μm external diameter in subjects with very severe emphysema has been shown to correlate with the pulmonary arterial pressure during exercise or with the difference between pulmonary artery pressure at rest and during exercise,11 suggesting that this type of remodelling leads to reduced distensibility of the pulmonary vessels. There are, however, conflicting data with regard to the amount of muscle in the pulmonary arterial wall. Many studies have failed to identify any abnormalities or found only minimal increases in wall thickness, even in the lungs of patients with severe COPD.7,10 Others have identified medial thickening in the vessels of smokers compared with non-smokers,12 in addition to a clear progression in thickness of the muscular wall from non-smokers to smokers without airflow obstruction to those with airflow obstruction, accompanied by an increase in the proportion of smaller sized vessels.8 However, muscular medial thickening, as opposed to overall vascular wall thickening, does not appear to have any clear relationship with the severity of pulmonary hypertension or with the ability of the vasculature to respond to oxygen.9
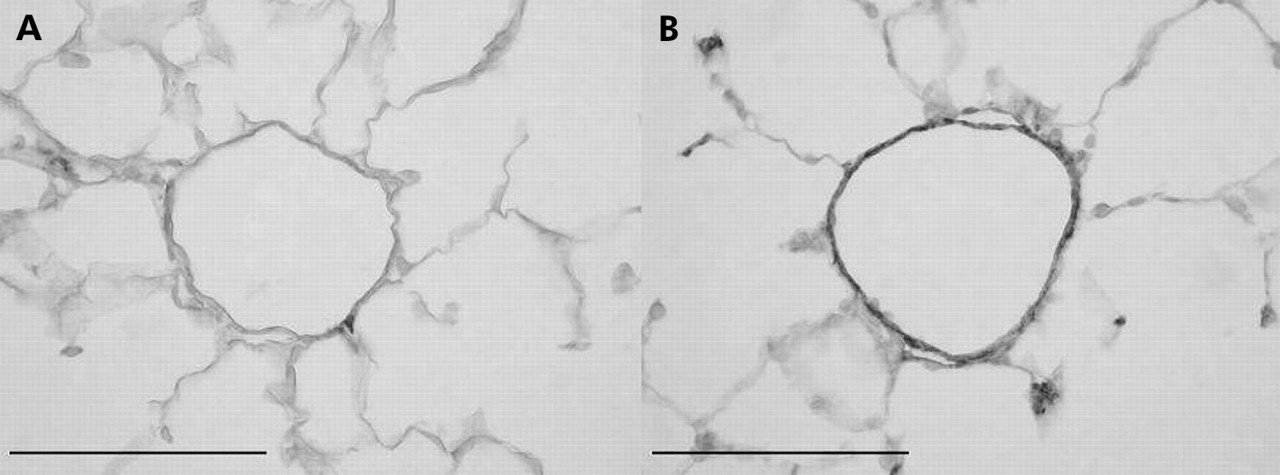
In patients with severe COPD there is a consistent finding of muscularisation of the (normally poorly muscularised) arterioles, consisting of both a circular and longitudinal muscle coat often accompanied by longitudinal tube formation, the latter indicating aberrant remodelling. Animal models of cigarette smoke exposure have clearly shown increased muscularisation of the partially muscularised arteries adjacent to the alveolar ducts (fig 1),6,13 and these studies have suggested that this does not regress upon cessation of smoking.

Smooth muscle actin stains illustrating muscularisation of the small, normally partially muscularised arteries adjacent to the alveolar ducts in female guinea pigs exposed either to room air (A) or cigarette smoke (B) for 6 months. The vessel from the control animal (A) has only a few slips of smooth muscle while that from the smoke exposed animal (B) is almost totally muscularised. Similar changes have been described in the lungs of humans with COPD. Magnification bar = 100 μm.
Very few studies have examined the adventitial compartment of the muscular arteries and no consistent changes have been found.7,9,10 An exception, however, is that arteries in smokers, both with and without COPD, appear to have an increased number of adventitial CD8 lymphocytes.10 These findings imply that structural reorganisation of the vessels—with increased muscularisation of the muscularised and partially muscularised small arteries—may be a direct consequence of cigarette smoking, that in humans there is considerable variability in this response, and that this response may precede the vascular physiological alterations of COPD. However, the implication of these reports, taken in aggregate, is that increased muscularisation in itself does not provide a consistent explanation for the presence of pulmonary hypertension in patients with COPD.
AETIOLOGY OF CIGARETTE SMOKE INDUCED PULMONARY HYPERTENSION
Hypoxia and emphysema
The early literature suggested that destruction of the pulmonary vascular bed by emphysema led to pulmonary hypertension. However, published studies have failed to show direct correlations between the pulmonary arterial pressure and emphysema, or between emphysema and right heart weight, to support this claim.14,15 This lack of correlation has also been seen in animal models in which chronic smoke exposure produces emphysematous airspace enlargement with decreased capillary density and capillary narrowing, but these changes show no relationship to increases in pulmonary arterial pressure.
Hypoxia, such as is found at high altitude, is certainly known to induce pulmonary hypertension. Although there is often co-existence of pulmonary hypertension and hypoxaemia in patients with severe emphysema, it seems clear that—other than in states of severe hypoxia—hypoxia alone is not the primary driver of vascular reorganisation and pulmonary hypertension, and arterial oxygen tension (Pao2) has not been identified as an independent predictor of mean pulmonary arterial pressure.14 In addition, despite a widespread belief that the presence of pulmonary hypertension in emphysema reflects loss of capillary bed and/or hypoxia secondary to emphysema, pulmonary hypertension in fact appears to be independent of hypoxia since, in patients without severe hypoxaemia (Pao2 >7.3 kPa), mean pulmonary arterial pressure was identified as the most significant prognostic factor. In patients with severe airflow obstruction requiring long term oxygen therapy, a mean pulmonary arterial pressure of more than 25 mm Hg reduced the 5 year survival to 36.3% compared with 62.2% when the pulmonary arterial pressure was less than 25 mm Hg.16 Furthermore, a clinical trial evaluating oxygen administration to patients with no to mild pulmonary hypertension in a setting of mild to moderate hypoxaemia, as well as further desaturation during sleep, did not alter the pulmonary haemodynamics over the 2 year follow up period. Finally, administration of nightly oxygen to subjects with severe hypoxaemia did decrease pulmonary arterial pressure, but pathological analysis of the lungs from these patients showed vascular abnormalities that were consistent from patient to patient and thus could not explain the varying physiological responses to exercise, nor the vascular response to oxygen seen in some patients.9
Dynamic changes in the pulmonary vessels
Another paradigm suggests that pulmonary hypertension is a result of dynamic changes in the pulmonary vascular bed. This includes two general postulates that are not mutually exclusive. The “structural” theory proposes that early morphological changes in the vessels do occur, but initially do not interfere with vessel dilatation during times of increased flow. However, when the structure becomes so altered that the vessel is non-compliant, increased flow results in increased pulmonary arterial pressure.7 In support of this hypothesis, a decrease in pulmonary artery compliance after chronic smoke exposure has been reported in a rat model.17 Alternatively, it has been suggested that the compliance of the vessels can be affected by changes in pulmonary function, with gas trapping causing vessel compression.7,18 Indeed, pulmonary arterial pressure does appear to correlate with the degree of airflow obstruction, even in patients with mild airflow limitation.7 However, when vascular dynamics have been measured after lung volume reduction surgery, a technique which should reduce vessel compression by gas trapping, there was no change in pulmonary arterial pressure either at rest or during exercise, suggesting that either very small amounts of gas trapping have a major effect or that, in subjects with this severity of disease, the vessels are already rigid due to altered structure and gas trapping does not exert an additive effect. Nevertheless, this concept has been supported by the sole animal study in which it was examined;6 in guinea pigs, physiological evidence of airflow obstruction was accompanied by increases in pulmonary arterial pressure while animals with a similar degree of emphysema but only mild alterations of airflow had no significant increases in pulmonary arterial pressure.
Endothelial dysfunction and vascular remodelling
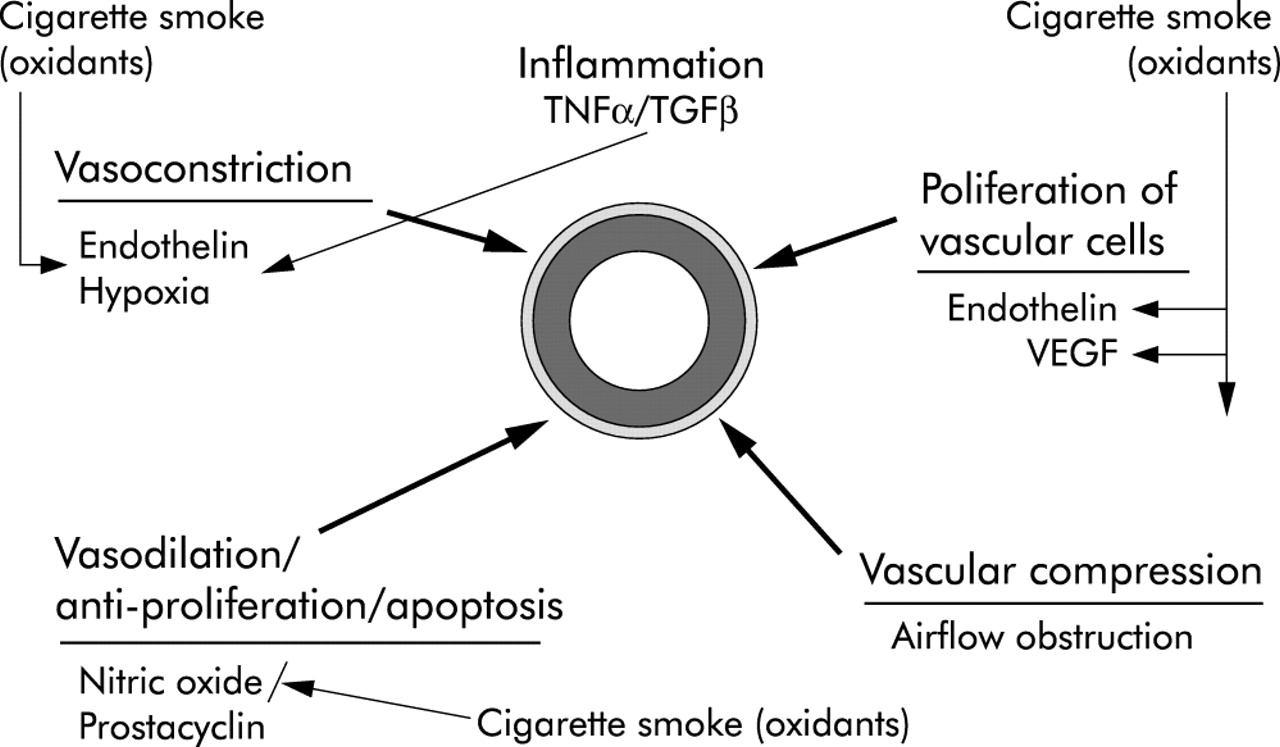
In most forms of non-COPD related pulmonary hypertension, the nature of the initial stimulus that causes vascular remodelling is unknown although injury to endothelial cells is frequently postulated. “Endothelial dysfunction” is a general term for a variety of abnormalities in endothelial cell structure and/or mediator production. The endothelial dysfunction theory19 posits that there is a regulatory disturbance in the production of, or the response to, mediators of tone and/or cellular replication, thus ultimately leading to vascular remodelling and aberrant vascular physiology (fig 2). Because most of these mediators affect smooth muscle growth and proliferation, endothelial dysfunction is believed to be one force driving the vascular hypertrophy and abnormal vascular structure seen in patients with pulmonary hypertension. In addition, endothelial injury may produce loss of the endothelial barrier, allowing circulating mediators and growth factors to directly affect the underlying vascular wall.
{kind=link}
{kind=link}
Diagram showing various cigarette smoke related processes that drive vasconstriction, vasodilation, and cell proliferation in the walls of the small pulmonary arteries. Most of the effects shown are potentially directly mediated by oxidants in the smoke acting on the vessel wall. The vessel is shown in the centre with muscular layer and adventitial layers highlighted. Oxidants in the smoke cause production of the vasoconstrictor endothelin and the vasoproliferative agents endothelin and vascular endothelial growth factor (VEGF). Smoke interferes with nitric oxide mediated vasodilation by decreasing nitric oxide bioactivity. Air trapping as part of chronic airflow obstruction appears to play an early role by inducing vascular compression, and hypoxia causes vasoconstriction, but only when the oxygen tension is extremely low.
In smokers, endothelial dysfunction and remodelling might result from the direct action of cigarette smoke on the vessel, acting to alter cytokine or vasoactive mediator balance.13 It could also be caused by the consequences of an inflammatory reaction in the vessel wall.10 Such dysfunction has been identified in the extrapulmonary pulmonary arteries of patients with COPD where there is a loss of nitric oxide dependent relaxation.20 This effect was not the result of abnormal membrane receptors or of l-arginine deficiency, but appeared to be associated with intimal thickening.
There is a large literature on the effects of endogenous mediators including vasodilators such as nitric oxide (NO) and the lipid mediator prostacyclin, vasoconstrictors such as endothelin, and the mitogenic stimulator vascular endothelial growth factor (VEGF) in pulmonary arterial hypertension of other aetiologies.21,22
Endothelium derived NO is believed normally to act as a vasodilator and has been shown to inhibit smooth muscle proliferation as well as downregulating vascular endothelin production.23 It is produced by the activity of endothelial nitric oxide synthase (eNOS) and both shear stress and increased pulmonary blood flow appear to modulate eNOS activity. In mice, overexpression of eNOS protects against hypoxic pulmonary hypertension,24 whereas eNOS knockout mice develop pulmonary hypertension with exposure to only mild hypoxia.25
Prostacyclin is a vasodilator that also protects against vascular remodelling and pulmonary arterial hypertension. While prostacyclin overexpression protects mice against hypoxia induced pulmonary hypertension,26 prostacyclin receptor deficient mice develop severe pulmonary hypertension when placed in a hypoxic environment.27 Decreased levels of prostacyclin synthase have been found in the pulmonary arteries of humans with various forms of pulmonary arterial hypertension and treatment with intravenous prostacyclin (epoprostenol) improves haemodynamics and survival, as well as decreasing circulating endothelin and increasing VEGF levels.28
Endothelin-1 (ET-1) is a powerful endothelium derived vasoconstrictor peptide. Endothelin levels are raised in experimental hypoxia and monocrotaline induced pulmonary hypertension as well as in humans with pulmonary arterial hypertension, and ET-1 levels correlate with pulmonary vascular resistance in such patients.29 ET-1 receptor antagonists produce haemodynamic and clinical improvement in patients with primary pulmonary arterial hypertension.30
Although endothelial cells in pulmonary hypertension produce increased immunoreactive VEGF, the role of VEGF in pulmonary hypertension is not entirely clear. In rats, chemical VEGF receptor blockade produces severe pulmonary hypertension31 and delivery of VEGF by viral vector protects against hypoxic pulmonary hypertension.32 In normal aortic endothelial cells VEGF induces production of prostacyclin and NO,33 thus favouring vasodilation. However, it has been suggested that, in the setting of vascular damage, VEGF potentiates damage by acting as a growth factor. VEGF also induces matrix metalloproteinase-1,34 an effect that may contribute to vascular remodelling (see below).
There are few studies that have examined these mediators in human cigarette smokers and patients with COPD, and the results reported are not entirely consistent. Reports based on immunohistochemical approaches have found apparent reductions in eNOS staining in the intrapulmonary arteries of smokers accompanied by a decrease in eNOS protein in bulk lung samples.10 Increased endothelin immunoreactivity has been identified in the pulmonary vessels in a limited number of patients with pulmonary hypertension secondary to COPD.29 In addition, increased circulating endothelin levels have been found in cigarette smokers and in patients with COPD.35 Interestingly, arterial immunoreactive staining, bulk lung protein, and mRNA levels of VEGF were found to be increased in smokers without COPD and in smokers with moderate COPD, but decreased in the lungs of those with severe emphysema.36 There are no data on prostacyclin levels in cigarette smokers.
A number of studies have looked at production of these mediators in experimental systems. In an endothelial cell culture model, cigarette smoke induced endothelin production and reduced eNOS production and activity.37 Using bulk lung or more sensitive microdissection methods which allow specific evaluation of gene expression levels in small intrapulmonary arteries, rapid increases in endothelin and VEGF mRNA levels and immunoreactive protein have been found in animals acutely exposed only once to smoke,38 and this upregulation was sustained for up to 6 months with chronic exposure13 (Wright J et al, unpublished data). In contradistinction to the human data described above, eNOS mRNA and immunoreactive protein were also increased in these studies. Thus, while the data are relatively consistent for VEGF and endothelin in humans and animals, the eNOS data are discrepant.
Nonetheless, these findings strongly suggest that cigarette smoke directly induces the production, within the arterial walls, of vasoactive mediators which control dynamic vasoconstriction and vasodilation. While these effects are somewhat stereotypic in inbred strains of animals in the sense that every animal shows a fairly similar response, in outbred Hartley strain guinea pigs, only some of which develop pulmonary hypertension after chronic cigarette smoke exposure, there is marked variability in the level of gene expression of these mediators,13 and levels of mediator production directly correlate with the pulmonary artery pressure in guinea pigs exposed to smoke for 6 months (Wright J et al, unpublished data). This observation again implicates intrinsic effects of smoke on the vasculature as direct drivers of vascular remodelling and pulmonary hypertension, and also raises the possibility that genetic polymorphisms controlling levels of expression of these mediators may be important in determining which human smokers develop pulmonary hypertension.
Cell proliferation and vascular remodelling
Cellular proliferation has not been measured in the pulmonary vasculature of human smokers. In the larger (∼450 μm diameter) pulmonary arteries of rats exposed to smoke for up to 3 months, there was an increase in the volume proportion of fibroblasts, collagen bundles, and elastic laminae and a decrease in smooth muscle cells.17 In the smaller muscular vessels adjacent to the bronchioles and in the partially muscularised small arteries adjacent to the alveolar ducts, acute in vivo smoke exposure was associated with cell proliferation, and this proliferation was maintained during chronic smoke exposure of several months duration.13 In a lung slice explant model of acute smoke exposure, cell proliferation could be significantly reduced by administration of endothelin antagonists, endothelin converting enzyme inhibitors, and glutathione, the latter suggesting a potential role for oxidants in the induction of cell proliferation. These findings again support a direct effect of smoke on vascular remodelling, effects that are independent not only of airflow obstruction but also of smoke induced inflammation.
Proteases and vascular remodelling
To our knowledge, no studies have examined protease involvement in the vascular remodelling found in human COPD, but animal models of various (non-smoke) forms of pulmonary hypertension have clearly demonstrated a role for proteases in facilitating vascular remodelling.21 Exposure to hypoxia or monocrotaline causes expression of an endogenous serine elastase in the pulmonary arteries in rats,21 and elastase inhibition protects against pulmonary hypertension as well as vascular structural changes in these models. The role of endogenous serine elastase is believed to be activation of a variety of matrix metalloproteases, which themselves may cause vascular remodelling as well as the release of growth factors from matrix resulting in cell proliferation. Serine and metalloelastases also induce expression of tenascin-C,21 a substance that causes growth factor receptor phosphorylation and enhances the response to other growth factors. These studies indicate that both serine and metalloproteases are integral to the vascular remodelling process.
A single animal model has examined the effect of administration of an oral serine elastase inhibitor on vascular structure in animals exposed to cigarette smoke and found a significant decrease in the amount of small vessel cell proliferation and muscularisation.39 We have observed upregulation of gene expression of various matrix metalloproteases in the small intrapulmonary arteries in such animals (Wright J, Churg A, unpublished data).
IMPLICATIONS FOR TREATMENT
It is interesting to note that, despite substantial evidence against the notion that pulmonary hypertension in COPD is attributable simply to hypoxia and/or destruction of the pulmonary vascular bed by emphysema, COPD related pulmonary hypertension tends to be lumped with the pulmonary hypertension seen in other primarily ventilatory conditions from a classification and treatment standpoint. In fact, the classification of pulmonary hypertension developed at the World Health Organization international conference and recently updated at the Third World International Conference in Venice in 2003,22 puts pulmonary hypertension in COPD under the heading “Pulmonary hypertension associated with hypoxemia”, a grouping which includes COPD, interstitial lung disease, sleep disordered breathing, alveolar hypoventilation disorders, and chronic exposure to high altitude.40 Unfortunately, this generally useful classification does not take into consideration the extensive evidence outlined above which indicates that pulmonary hypertension is probably not related simply to hypoxia in COPD, or new mechanistic data that cigarette smoke has a direct effect on the pulmonary vasculature with upregulation of mediators that lead to vascular structural remodelling and dynamic physiological changes in vascular function.
Recognition of these direct smoke induced vascular pathophysiological processes should lead to the more active pursuit and investigation of specific treatments directed towards their abrogation or amelioration. This could result in a reduction in the consequences of pulmonary hypertension in cigarette smokers and subjects with COPD, not just in terms of mortality but also by improving functional capacity, exercise tolerance, and health related quality of life. Although currently available pulmonary hypertension specific treatments such as prostanoids, endothelin receptor antagonists, and phosphodiesterase inhibitors are expensive, they may in fact provide important beneficial effects for patients with COPD and result in an overall decreased cost to the healthcare system.
In summary, although the pulmonary vasculature is affected by airflow obstruction and probably by severe emphysema and hypoxia, the traditional views that emphysema and hypoxia explain the presence of pulmonary hypertension in COPD are not really supported by the data. There is evidence that, in patients with severe hypoxia, administration of oxygen at night is beneficial, but this approach is not of value in most patients with COPD and pulmonary hypertension. It is time to take a fresh look at the implications and potential treatment of pulmonary hypertension in COPD based on new pathophysiological insights.
REFERENCES
Footnotes
-
This study was supported by grant MOP 62693 from the Canadian Institutes of Health Research. The work is independent of the funders.
-
Competing interests: JLW and AC have received funds from AstraZeneca to examine animal models of emphysema and to test the effectiveness of various compounds under development. RDL has served on advisory boards for Actelion in 2002 ($1000 CAD), 2003 ($2500 CAD) and 2004 ($1200 CAD), has served as a consultant to GlaxoSmithKline ($3375 CAD in 2003) and Northern Therapeutics ($1750 CAD in 2004), and has received fees from Actelion for lecturing at continuing health education activities ($2000 CAD in 2002; $750 CAD in 2003; $1250 in 2004) and $34 000 CAD from Encysive, LP as a research grant for participating in a multicentre clinical trial in 2003–4.