Article Text

Abstract
Background: The inflammatory process in chronic obstructive pulmonary disease (COPD) is characterised by the presence of neutrophils in the lung that are able to synthesise de novo several inflammatory mediators. The local chronic persistent inflammatory response is accompanied by systemic effects such as cytokine induced priming of peripheral leucocytes and muscle wasting. The preactivation or priming of peripheral blood neutrophils was used to gain more insight into the mechanisms of this systemic inflammatory response.
Methods: Gene arrays were performed on peripheral blood neutrophils obtained from healthy donors after stimulation in vitro with tumour necrosis factor (TNF)-α, granulocyte-macrophage colony stimulating factor (GM-CSF), or both. The expression of many inflammatory genes was regulated in these cells following stimulation. The expression of inflammatory genes in peripheral blood neutrophils in healthy subjects and those with COPD was measured by real time RT-PCR after stimulation with TNFα, GM-CSF, interleukin (IL)-8, fMLP, TNFα + GM-CSF, and lipopolysaccharide (LPS).
Results: The genes regulated in the gene array with TNFα/GM-CSF stimulated neutrophils included cytokines (such as IL-1β), chemokines (such as IL-8), and adhesion molecules (such as ICAM-1). Disease severity as measured by forced expiratory volume in 1 second (FEV1) in COPD patients correlated with expression of several of these genes including IL-1β (r = −0.540; p = 0.008), MIP-1β (r = −0.583; p = 0.003), CD83 (r = −0.514; p = 0.012), IL-1 receptor 2 (r = −0.546; p = 0.007), and IL-1 receptor antagonist (r = −0.612; p = 0.002).
Conclusions: These data are consistent with the hypothesis that progression of COPD is associated with the activation of neutrophils in the systemic compartment. De novo expression of inflammatory mediators by peripheral blood neutrophils suggests a pro-inflammatory role for these cells in the pathogenesis of COPD.
- chronic obstructive pulmonary disease
- neutrophils
- systemic inflammation
- gene profiling
Statistics from Altmetric.com
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease of the airways which is characterised by the presence of chronic airflow obstruction and an abnormal inflammatory response in the pulmonary tissue.1,2 The aberrant inflammatory process is characterised by increased levels of cytokines and/or chemokines such as tumour necrosis factor (TNF)-α and interleukin (IL)-8.3,4 Furthermore, an increase in neutrophils, monocytes, and CD8+ cytotoxic lymphocytes is found in the pulmonary compartment.5,6 This increase in inflammatory cells is likely to be mediated by the increased levels of cytokines and/or chemokines in the tissue. In addition, the presence of these cytokines in the plasma/serum of patients with COPD strongly suggests that the local inflammatory response communicates via these mediators with the systemic circulation.7,8,9,10
Systemic effects are initiated by the persistent local inflammatory response in COPD patients. In contrast to the local inflammation, little is known concerning the systemic inflammatory response in COPD. The identification of soluble TNF receptors (which are thought to counteract the function of TNFα) in the plasma of patients with COPD corroborates the idea that the local inflammation in the airways communicates with the systemic compartment.4 Unfortunately, direct measurement of many inflammatory mediators such as TNFα is difficult in the peripheral blood because of their low concentration, short half life, binding to soluble receptors, and renal clearance.11 Alternatively, a few studies have reported that neutrophils in the peripheral blood of COPD patients exhibit characteristics of a primed phenotype.12,13 These findings suggest that the activation of neutrophils may start in the circulation. However, the underlying pathogenetic mechanisms remain to be determined.
New methods that allow monitoring of neutrophil activation and analysis of inflammatory mediators are important for the characterisation of the systemic inflammatory response in COPD. We and others have shown that in vitro priming of neutrophils with, for example, granulocyte-macrophage colony stimulating factor (GM-CSF) is associated with the expression of specific gene profiles.14,15 The investigation of gene expression profiles of peripheral blood neutrophils of COPD patients in comparison with in vitro cytokine induced gene profiles of neutrophils could therefore be a powerful tool for characterising chronic systemic inflammation in COPD.
In this study peripheral blood neutrophils of healthy donors stimulated with TNFα, GM-CSF, or both were examined by analysis of cytokine regulated gene profiles in vitro using gene arrays. Further investigation of differentially expressed genes such as several IL-1 family members, macrophage inflammatory protein (MIP)-1β, and CD83 in patients with mild and severe COPD showed that the expression of these genes in peripheral blood neutrophils correlates with the severity of COPD.
METHODS
Reagents
fMLP and LPS (E coli) were purchased from Sigma (St Louis, MO, USA), recombinant human GM-CSF from Genzyme (Boston, MA, USA), recombinant human TNFα from Boehringer Mannheim (Germany), IL-8 from Peprotech (Rocky Hill, NJ, USA), human serum albumin (HSA) was obtained from the Central Laboratory of the Netherlands Red Cross Blood Transfusion Service (Amsterdam, The Netherlands), RPMI 1640 medium with glutamax was purchased from Life Technologies (Breda, The Netherlands), and Ficoll-Paque was from Pharmacia (Uppsala, Sweden).
Preparation of samples
Blood was obtained from healthy volunteers and COPD patients. Granulocytes were isolated as described previously.16 Neutrophils used for in vitro stimulation experiments as well as for RNA isolation were incubated in RPMI 1640 glutamax supplemented with 0.5% HSA for 15 minutes before stimulation or further preparation. RNA isolation and cDNA synthesis were performed as previously described.16 All preparations contained >97% neutrophils, and contaminating cells were mainly eosinophils.
Subjects
Twenty three patients with clinically stable moderate to very severe COPD were included in the study. COPD was defined according to the criteria of the GOLD guidelines.1,2 Inclusion criteria were forced expiratory volume in 1 second (FEV1) <80%, reversibility of less than 10% or less than 200 ml after inhalation of a β2 agonist, and a FEV1/FVC ratio of less than 70% of the predicted value. Subjects with a history of other concomitant confounding diseases such as diabetes mellitus, lung carcinoma, thyroid and cardiovascular disease and those with bronchiectasis were excluded from the study. Patients with an exacerbation due to a respiratory tract infection or other respiratory complaints during the 4 weeks before the study or those with a history of asthma or atopy were also excluded. Eleven patients were current smokers while the rest had stopped smoking at least 1 year before the study. Details of the patients are shown in table 1.
Characteristics of study patients
Fourteen of the 23 patients met the criteria for moderate COPD (class II according to the GOLD guidelines) while the other nine had severe to very severe COPD (class III and IV). All patients were treated with inhaled long acting β2 agonists (salmeterol 50 μg twice daily), inhaled corticosteroids (250 μg twice daily), and additional bronchodilating agents if needed. Regular use of oral glucocorticosteroids was an exclusion criterion. Eleven healthy never smokers were enrolled as controls, all of whom had normal lung function and no medical history of pulmonary diseases.
The study was approved by the medical ethics committee of the University Medical Center, Utrecht and informed consent was obtained from all subjects.
Gene array analysis
Isolated human neutrophils were incubated in RPMI 1640 supplemented with 0.5% HSA for 15 minutes and stimulated for 3 hours at 37°C in the presence or absence of cytokines, followed by RNA isolation as described previously. RNA was treated with DNase to avoid contamination with genomic DNA and subsequently analysed by agarose gel electrophoresis to verify the integrity of the RNA preparations. PolyA+ RNA was extracted with magnetic beads coated with poly dT using the Atlas Pure Total RNA Labeling System (Clontech, Palo Alto, CA, USA) according to the manufacturer’s protocol. 40 μg of total RNA from neutrophils was used for each gene array. The gene array screen was performed with the Atlas Human Hematology/Immunology Array #7737-1 (Clontech) using reagents supplied and the manufacturer’s protocol. Briefly, cDNA probes were generated using specific primers for the genes on the array. Reverse transcription of the isolated polyA+ RNA was performed with radioactive γ32P-dATP. The two arrays were incubated overnight with the probe at 68°C. The following day the arrays were washed and membranes analysed by autoradiography. The spots on the arrays were quantitated using a Phosphor Imager and ImageQuant software (Amersham Biosciences, Uppsala, Sweden). Values were corrected for background and housekeeping genes.
Real time PCR
Gene expression was analysed by real time RT-PCR. For CD83 the Taqman probe procedure was used; all other genes were analysed with SYBR green I. For β-actin we used primers as described previously.17 All other primers were designed using Primer 3 software from the Whitehead Institute/MIT Center for Genome Research (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) and are shown in table 2. The results were normalised for the housekeeping gene β-actin and GAPDH. A reference sample of cDNA on every 96-well plate allowed correction of differences between plates.
Sequences of primers used for real time PCR (SYBR green)
Statistics
Statistical analysis was performed with SPSS 10.0. Multiple comparisons where analysed by analysis of variance (one way ANOVA, Fisher test). In addition, the results were analysed using the Mann-Whitney U test. For the analysis of the data depicted in fig 1 a paired t test was used. Correlation coefficients were calculated using Spearman’s correlation coefficient. Statistical significance was defined as p<0.05.

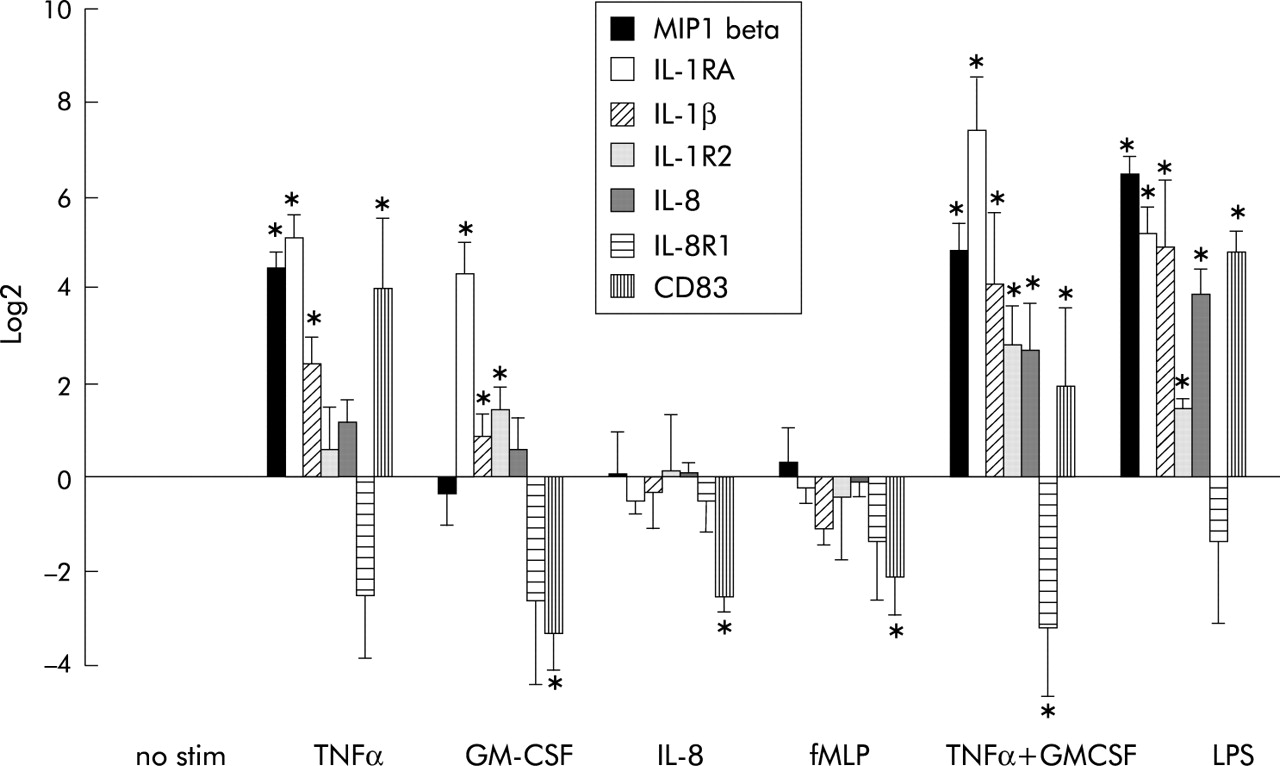
Gene expression in peripheral blood neutrophils stimulated by several mediators in vitro. Peripheral blood neutrophils were incubated with several physiological stimuli for 3 hours in vitro at 37°C (TNFα, 100 U/ml; GM-CSF, 0.1 nM; IL-8, 10 nM; fMLP, 1 μM; LPS, 10 ng/ml). Expression of indicated genes was measured by real time RT-PCR. The results are expressed as mean (SD) log2 fold change (n = 3). Differences between values were compared by the paired t test. *p<0.05.
RESULTS
Modulation of inflammatory genes in neutrophils by TNFα and/or GM-CSF
To gain further insight into the mechanisms of neutrophil activation, we evaluated differential gene expression in healthy donor blood neutrophils activated with GM-CSF and TNFα in vitro. Table 3 shows the change after stimulation of neutrophils with the cytokines TNFα or GM-CSF separately or in combination compared with non-stimulated neutrophils. Most of the genes on the array encoded for cytokines/chemokines and cell surface receptors. Among the cytokines and chemokines, genes encoding for IL-8, IL-1β, and IL-1 receptor antagonist (IL-1RA) were clearly upregulated by both TNFα and GM-CSF. On the other hand, more specific signals induced by the cytokines were shown by the regulation of MIP-1β which was only induced by TNFα and by lymphotoxin-β which was only induced by GM-CSF. In addition, this analysis revealed that TNFα and GM-CSF both induced pro-inflammatory genes such as IL-1β and IL-8, as well as anti-inflammatory genes such as IL-1RA and IL-1 receptor 2 (IL-1R2). Of the regulated cell surface antigens, most were upregulated by both TNFα and GM-CSF except for CD83 and CD69 which were only upregulated by TNFα or GM-CSF, respectively. The anti-apoptotic Bcl-2 family member A1 is involved in regulating cell survival and expression was already high directly after isolation, decreasing after 3 hours at 37°C in the absence of cytokines. Both TNFα and GM-CSF maintained the high expression of Bcl-A1.
Genes regulated by several stimuli in neutrophils of healthy donors (n = 2)
The combination of TNFα and GM-CSF resulted in regulation of almost all genes which are regulated by TNFα and GM-CSF individually, but some genes were regulated only by the individual cytokines. MIP-1β, CD83, IL-8R1, and antileukoproteinase (ALP) were only regulated by TNFα, whereas GM-CSF alone regulated retinoic acid receptor (RAR)-α, B cell translocation gene 1 (BTG1), lymphotoxin-β, CD69, MNDA, STAT5A/B, and vav2 (table 3).
The expression of MIP-1β, IL-1RA, IL-1β, IL-8, IL-8R1, and CD83 regulated on the gene array (see table 2) was confirmed by real time RT-PCR. Peripheral blood neutrophils of three healthy donors were stimulated with TNFα, GM-CSF, IL-8, fMLP, and LPS and gene expression was measured with real time RT-PCR (fig 1). Analysis revealed that IL-8 and fMLP did not induce any of the genes investigated except CD83. Upregulation of the mRNA for IL-1RA and IL-1β was observed after stimulation of the cells with TNFα, GM-CSF, LPS, or co-stimulation with TNFα and GM-CSF. IL-1R2 was downregulated by all stimuli. Similar to the data obtained with the gene arrays, CD83 and MIP-1β were upregulated by TNFα and not by GM-CSF.
Expression of TNFα and GM-CSF regulated genes in COPD patients
Real time RT-PCR analysis of gene expression in COPD patients and healthy controls is shown in fig 2. In neutrophils of patients with moderate COPD (GOLD class II) only the IL-8R1 gene was significantly decreased compared with healthy volunteers. In patients with more severe COPD a significant increase was seen in CD83, MIP-1β, and IL-1β compared with healthy volunteers. Furthermore, CD83, IL-1β, MIP-1β, and IL-1RA were significantly increased in patients with severe and very severe COPD compared with those with moderate COPD.

Gene expression analysis in peripheral blood neutrophils of COPD patients. Peripheral blood neutrophils were isolated from healthy volunteers (filled squares), moderate (class II) COPD patients (open diamonds), and those with severe to very severe (class III and IV) stable COPD (filled triangles). Expression of indicated genes was measured using real time RT-PCR and expressed as log2 fold change compared with healthy controls. Differences between the study groups were compared using the one way ANOVA, Fisher test.
TNFα regulated genes correlated with severity of disease in patients with stable COPD. CD83 (r = −0.51), MIP-1β (r = −0.58), IL-1RA (r = −0.61), IL-1β (r = −0.54) and IL-1R2 (r = −0.546) correlated with the severity of COPD as measured by FEV1 (fig 3). In contrast, there was no correlation between FEV1 and the expression of IL-8 and IL-8R1 (results not shown). Current smoking status did not influence the gene expression in fig 3 and BMI did not correlate with severity of disease in patients with stable COPD (results not shown).

{kind=link}
{kind=link}
{kind=link}
Correlation of gene expression in peripheral blood neutrophils of COPD patients with FEV1 (% predicted). Expression of indicated genes was measured using real time RT-PCR and expressed as log2 fold change compared with healthy controls. Correlations between FEV1 and expression of indicated genes in neutrophils from patients with stable COPD were evaluated using Pearson’s rank correlation analysis.
DISCUSSION
The chronic persistent inflammatory reaction in COPD patients is dominated by neutrophils which are effector cells that can also mediate the systemic effects of chronic inflammation.10,18 Investigation of gene expression in peripheral blood neutrophils of COPD patients could therefore provide a unique insight into the molecular mechanisms modulating chronic systemic inflammation. In this study the activation of peripheral blood neutrophils was examined by gene array analysis of cytokine stimulated neutrophils. mRNA expression of MIP-1β, IL-1RA, IL-1β, IL-8, IL-8R1, and CD83 in cytokine stimulated neutrophils and in neutrophils obtained from patients with COPD was measured with real time RT-PCR.
Gene expression analysis of peripheral blood neutrophils from patients with severe stable COPD indicated that peripheral blood neutrophils exhibited an activated phenotype. Clear regulation of pro-inflammatory mediators was evident in patients with severe to very severe COPD (class III and IV, fig 2); several of these genes including IL-1β, IL-1R and MIP-1β have previously been reported to be regulated by pro-inflammatory mediators such as TNFα, GM-CSF, or LPS.19–21 Upregulation of CD83, which is described as a specific marker for dendritic cells, has recently been reported to be expressed on neutrophils in vitro by TNFα and in vivo by acute infection.22,23 These data are consistent with the hypothesis that circulating neutrophils in patients with severe COPD have encountered pro-inflammatory mediators.
The genes regulated in the neutrophils of normal subjects stimulated in vitro with TNFα and LPS were very similar to the genes regulated in the patients with COPD. However, the gene expression profile of neutrophils from COPD patients is not identical to the profile of individual mediators such as TNFα or LPS. It is unlikely that dominant single mediators drive systemic inflammation in COPD but, rather, a combination of inflammatory mediators. Indeed, a combination of inflammatory mediators can lead to enhancement or inhibition of the transcription of genes (fig 1). A clear example is the finding that interferon (IFN)-γ enhances the production of cytokines/chemokines of neutrophils stimulated by LPS while IL-10 inhibits their production.20 In addition, the kinetics of gene expression could have an influence. Genes that are expressed relatively late upon activation might not be found in peripheral blood but only in cells that have migrated to the tissues.
Investigation of expression of several of these genes (including IL-1β, IL-1RA, IL-8, CD83 and MIP-1β) by real time RT-PCR in patients with different stages of COPD revealed that these genes were significantly increased in patients with more severe disease (class III and IV) and there was a significant negative correlation between the extent of gene expression in peripheral blood neutrophils and lung function. This was not a generalised finding since this correlation did not exist for several other genes (results not shown). These data show a correlation between lung function impairment and the systemic inflammatory reaction as measured in the peripheral blood. Systemic inflammation can thus be measured as an additional marker for the health status and may also help to determine the prognosis of a patient with COPD. The genes which correlated with lung function did not correlate with other parameters such as body mass index (BMI) or cotinine levels in the current smokers (results not shown). Low BMI is often a major health problem in smokers with COPD but, in our study, the BMI levels were not significantly different from healthy controls.
It has been shown that, besides the traditional functions such as phagocytosis, degranulation and production of superoxide, neutrophils can express a variety of inflammatory mediators (reviewed by Cassatella20). These findings suggest that circulating neutrophils not only participate in airway damage in COPD patients but also participate in the regulation of systemic inflammation.
TNFα, GM-CSF, and LPS can induce the expression of pro-inflammatory mediators in neutrophils and thereby enhance the inflammatory response. Natural inhibitory proteins such as IL-1RA and IL-1R2 inhibit IL-1β induced pro-inflammatory responses. IL-1RA exerts its inhibitory action by binding to IL-1 receptors without triggering any intracellular signalling responses, whereas IL-1R2 is a decoy receptor and binds IL-1β thereby competing for IL-1R expressed on inflammatory and/or bystander cells.24,25 Upregulation of IL-1RA and IL-1R2 in neutrophils in vivo is likely to be involved in dampening of the inflammatory response initiated by IL-1β. Similarly, TNFα or GM-CSF downregulated the expression of IL-8R1 which is likely to result in reduced responsiveness to this pro-inflammatory chemokine. Other studies have shown that systemic inflammation can cause functional downregulation of serpentine receptors in vitro and in vivo.26–28 Thus, activated neutrophils not only induce a pro-inflammatory response but also an anti-inflammatory response, generating a negative feedback loop. The outcome of these two processes may be determined by differences in the levels and kinetics of the peripheral pro- and anti-inflammatory cytokines. Neutrophils are therefore not only effector cells but might also have a role in the regulation of the duration of an inflammatory reaction.
An interesting observation is the disturbed balance of mRNA expression between the pro-inflammatory mediator IL-1β and the anti-inflammatory mediators IL-1RA and IL-1R2 in patients with severe stable COPD compared with in vitro where peripheral blood neutrophils are activated with inflammatory mediators. Real time RT-PCR analysis showed that the expression of IL-1RA is much higher than that of IL-1β in neutrophils stimulated with TNFα (fig 1) compared with neutrophils from patients with severe stable COPD (fig 2). The regulation of natural inhibitory genes is apparently disturbed in peripheral blood neutrophils from patients with severe COPD. In addition, expression of IL-1R2 is not upregulated in these patients while, in vitro, GM-CSF and LPS induced expression of IL-1R2. The balance of expressed genes has therefore been shifted towards a pro-inflammatory state.
In contrast to rheumatoid arthritis, little is known about the function of the IL-1 family of proteins in COPD. In the rheumatoid synovium an imbalance exists in the pro- and anti-inflammatory mediators since the relative levels of production of IL-1RA are not adequate to block the pro-inflammatory effects of IL-1 effectively.29 Injection of IL-1RA into the synovium significantly reduced the signs and symptoms of rheumatoid arthritis after 24 weeks.30 The imbalance between anti- and pro-inflammatory genes (including the IL-1 family) of circulating neutrophils might therefore also contribute to the development of a chronic inflammatory response in COPD.
Because of the relatively small sample size in this study, we could not evaluate putative correlations between measured variables and confounding factors such as age, circulating neutrophil number, or co-related smoking induced morbidity such as occult coronary artery disease. In a previous study performed by Malcolm et al,31 donor variability was observed by gene analysis in LPS stimulated neutrophils. We validated the genes regulated on the microarrays with real time RT-PCR in cytokine stimulated peripheral blood neutrophils of three healthy donors.
In the patients with COPD a single time point was studied. It would be interesting to study gene regulation during and after exacerbation of the disease, but this was beyond the scope of the present study. In addition, it would be interesting in future studies to compare cytokine induced gene profiles of other systemic inflammatory diseases such as rheumatoid arthritis with COPD.
In conclusion, this study indicates that activated neutrophils can be found in the peripheral blood of patients with stable COPD and this activation correlates with the severity of the disease. Inflammatory mediators such as TNFα are probably involved in the activation of neutrophils in these patients. Interestingly, there is an imbalance in the expression of pro- and anti-inflammatory mediators (including the IL-1 family) in the neutrophils of patients with more severe COPD. This imbalance towards a pro-inflammatory response may contribute to the systemic inflammation observed in these patients. Greater understanding of these mechanisms by fine tuning analysis of the gene profiles may increase our understanding of the systemic inflammation in COPD and may result in better markers for disease staging and prognosis.
REFERENCES
Footnotes
-
This work was supported by the Dutch Asthma Foundation (Grant 97.68) and ZonMW 9-40-37-035.
-
The first two authors contributed equally to the study.