Article Text

Abstract
Background:ADAM 33 is the first gene identified as a candidate for asthma by positional cloning techniques, with association studies reaching impressive statistical significance. It has a postulated role in myogenesis, airway modelling, and signalling via protein shedding. Concerns over the methodology of the initial study have led to several attempts at replication, with inconsistent results.
Method: To clarify the role of ADAM33 in determining the risk of asthma in the general population, new transmission disequilibrium and case-control studies were undertaken followed by a meta-analysis of all existing data.
Results: Studies in Icelandic and UK populations revealed no association when taken in isolation. The meta-analysis, however, showed that the F+1 and ST+7 variants were significantly associated with asthma in both types of study.
Conclusions: The additional risk imparted by this variation would account for 50 000 excess asthma cases in the UK alone. This study also demonstrates the size of study required to investigate such hypotheses adequately.
- asthma
- genetics
- ADAM33
Statistics from Altmetric.com
Asthma is a common but complex disease phenotype with both genetic and environmental factors contributing to its development. A study of 460 white families identified a candidate region on chromosome 20p by linkage analysis. A candidate gene within this region was identified by a positional cloning approach—namely, the membrane anchored disintegrin and metalloproteinase gene ADAM33.1 The metalloproteinase domains of such glycoproteins are mediators of intercellular and cell–matrix interactions via the proteolytic release of surface proteins (such as cytokines and receptors) and the cleaving of extracellular matrix components.2 The disintegrin regions support integrin mediated cell adhesion. These proteins have a role in fertilisation, myogenesis, and neurogenesis. The significance of ADAM33 in asthma is postulated to be related to airway modelling in utero and remodelling in later life, but the understanding of its physiological role is at an early stage.3 Recent advances include the localisation of ADAM33 to airway smooth muscle4 and preferential expression by airway fibroblasts of six alternative forms of ADAM33,5 all without the proteolytic domain.
Multiple single nucleotide polymorphisms (SNPs) in ADAM33 have been associated with asthma and bronchial hyperresponsiveness in both case-control and family linkage studies.6–9 Although results for single SNPs and haplotypes have in some studies reached impressive statistical significance (p values to 0.000003), the actual SNPs associated have varied between studies. This has raised the issue of multiple testing—for example, the paper by Van Eerdewegh et al1 contains over 700 analyses. To try to clarify the role of ADAM33 we have produced further data to expand the size of the informative populations and cumulatively analysed data from all published studies.
METHODS
Sixty nuclear families were recruited from an asthma outpatient clinic in Nottingham on the basis of pairs of affected offspring. The diagnosis of asthma was based on physician assessment. In addition, the majority underwent challenge testing with histamine and demonstrated bronchial hyperresponsiveness. Evidence of asthma symptoms and objective measures of airflow obstruction and reversibility were obtained for all parents and probands.
An additional 348 unrelated cases of asthma identified in the Icelandic population (selection outlined previously10) and 262 controls, in whom no evidence for asthma was present, were also genotyped to expand the case-control population. The controls were selected in part by being at least five meioses distant from each other. This population represents the largest case-control population studied for this gene. A separate cohort of 94 atopic non-asthmatic Icelandic subjects was also genotyped.
Both populations were genotyped at DeCode Genetics in Iceland. All patients signed an informed consent form. The study was approved by the Data Protection Authority, the National Bioethics Committee of Iceland, and stringent standards were followed to protect patient privacy.
Genomic DNA was isolated using a modified high throughput Qiagen isolation method as previously described.8 Genotyping was performed on the ABI7900 using a 5′ nuclease Taqman allelic discrimination assay (Assay-by-Design, Applied Biosystems, Denmark) in accordance with the manufacturers’ recommendations. The data were processed through an automatic allele calling system (less than 0.2% error rate in allele calling) as previously described.11 The Nottingham family data set was also checked for possible pedigree inconsistencies. The nomenclature of SNPs described in this paper follows that of previous work.
Previously performed case-control or transmission disequilibrium test (TDT) studies were identified from the literature by searching PubMed (www.ncbi.nlm.nih.gov/entrez/query.fcgi) for “ADAM33”. Numbers of cases and controls (or, in the TDT analysis, transmitted and non-transmitted allele frequencies) were calculated from these published studies: additional data were obtained from the authors of the GALA (Genetics of Asthma in Latin Americans) study6 to assist this analysis. SNPs for which data were available from three or more studies were included in the analysis. For both approaches this did not exclude any SNPs found to be associated with asthma. An initial analysis of allele frequencies reported in the GALA Hispanic populations showed no marked differences in distribution from the white ethnic populations, so data were pooled from this group to estimate population risk. A summary of the populations studied is shown in table 1.
Summary of populations studied
For each of the SNPs associated with asthma in the initial paper the odds ratio (OR) for asthma was calculated, given possession of the associated allele in each case-control study for which data were available. A weighted summary OR was derived by the Mantel-Haenszel method using the Clayton and Hills calculation of variance.
RESULTS
None of the SNPs under study was significantly associated with asthma in the Icelandic population alone. However, four of the 13 SNPs were significantly associated with asthma when all available data were combined in a meta-analysis (fig 1) with a maximum OR of 1.46 (95% CI 1.21 to 1.76) for ST+7 (p = 0.0001). There was no association between ADAM33 SNPs and atopy in the Icelandic population (data not shown). A χ2 test for heterogeneity found no evidence for effect modification by data source.

Odds ratios (with 95% confidence intervals) of asthma with ADAM33 polymorphisms shown; *p<0.05.
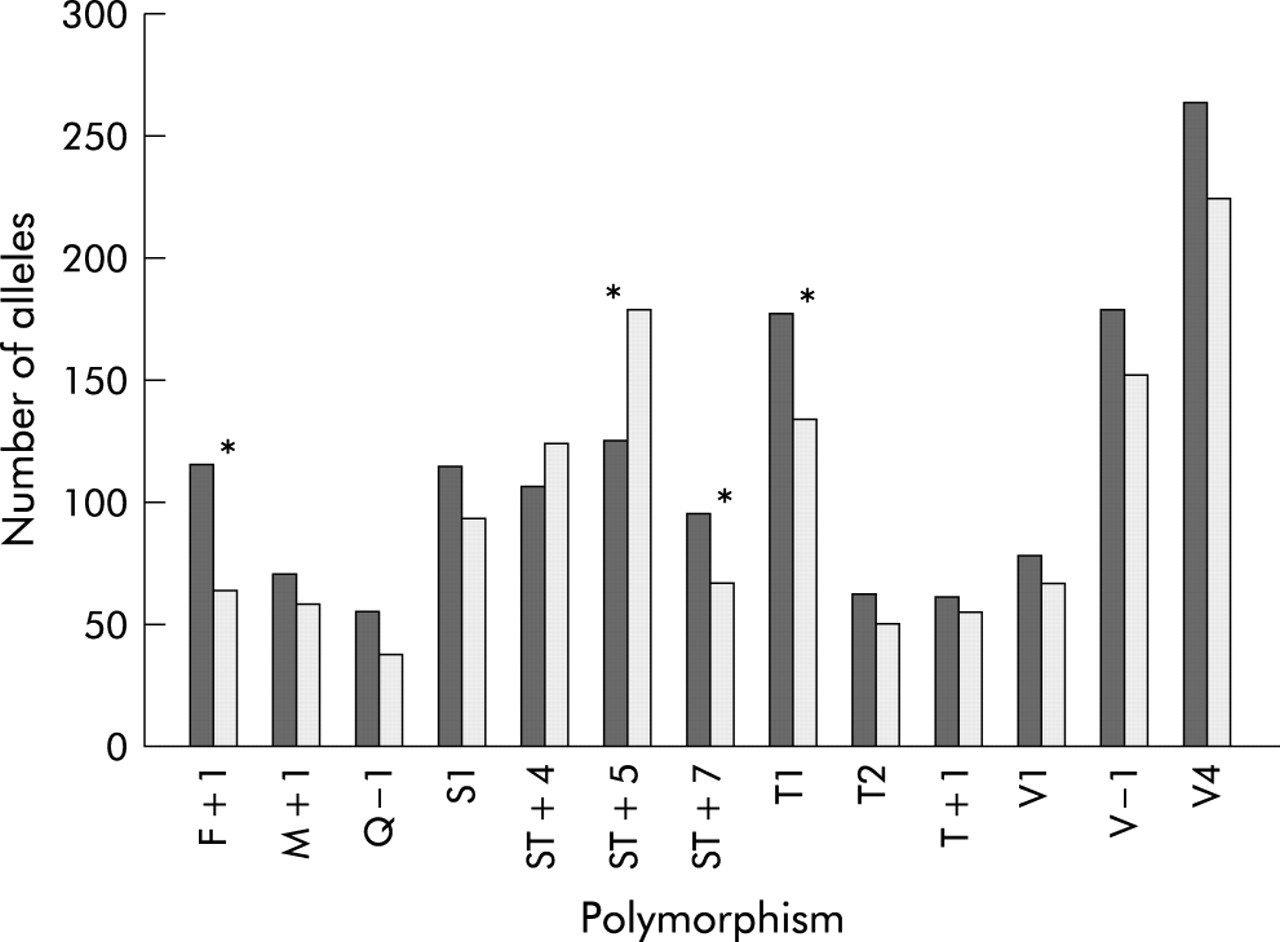
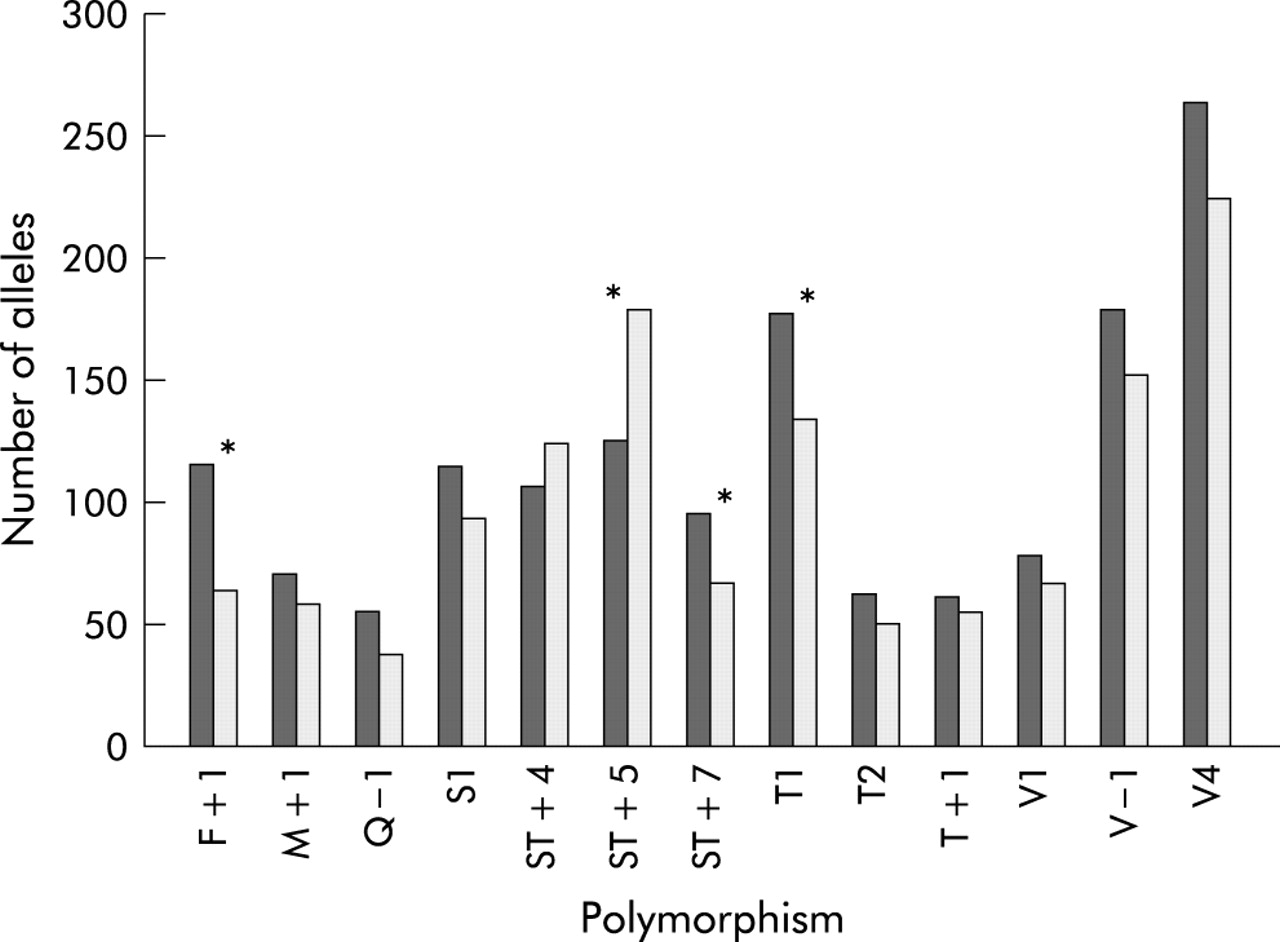
When a combined analysis was performed on all available TDT data, four SNPs were found to be transmitted significantly more commonly than predicted to subjects with asthma (fig 2), maximally F+1 (p = 0.002). This was despite the fact that no SNP was significantly overtransmitted in the Nottingham population alone.
{kind=link}
{kind=link}
Summary of TDT data. Alleles transmitted to asthmatics (dark bars) and those not transmitted (light bars); *p<0.05.
Thus, the F+1 and ST+7 polymorphisms were the only two to be significantly associated with asthma in both arms of the analysis. To clarify the extent of linkage disequilibrium between these markers, we used the Icelandic control population to estimate values of D′. F+1 and ST+7 are in tight linkage disequilibrium (D′ = 0.965).
DISCUSSION
Although the reported ORs seem to show a relatively small effect of these genotypes, their potential impact becomes clearer when considering the relevant allele frequencies. Using the figures from the overall analysis, the allele frequencies for ST+7 G was 84.9% in the asthmatic population and 79.1% in the control population. Based on an estimated asthma prevalence of 8%, this SNP would potentially contribute to around 50 000 excess asthma cases in the UK population. Although the precision of this estimate may be reduced due to the heterogeneity of doctor diagnosed asthma, ADAM33 is strongly associated with objective measures such as bronchial hyperresponsiveness1,9 and accelerated decline in forced expiratory volume in 1 second.12
Although the number of studies included in this analysis is relatively small, no definite evidence for publication bias is apparent. We also believe that this is unlikely to be a major problem with our analysis as the different published studies show positive associations with different SNPs.
One could argue that the initial positive study should be excluded from the meta-analysis because of publication bias and concerns over the methodology.9 If these data are excluded, the odds ratio for ST+7 is little changed at 1.4 (p = 0.0013). However, the odds ratio for F+1 is reduced to 1.167 (p = 0.1635), although this is now based on only two studies. Nonetheless, it is possible that the association with F+1 is solely due to linkage disequilibrium with ST+7.
There are a number of important implications of the new summarised data presented here. Firstly, this study suggests that the initial work on ADAM33 overestimates the importance of this locus at a population level. This is probably because the SNP analyses were only performed in families contributing to the linkage peak observed on chromosome 20. Secondly, our study confirms that relatively small case-control or TDT based approaches will be underpowered for determination of the contribution of loci such as ADAM33 to the asthma phenotype. Given the OR observed with ST+7, around 1500 cases and matched controls would be needed to identify this SNP in a single study with 90% power and p = 0.01. This lack of power is compounded by the common practice of examining the phenotypic association of one SNP in a study. We advocate initially including more SNPs to establish an association with the gene before undertaking more focused studies to identify SNPs upon which to concentrate functional studies. Finally, it is clear that several genes are likely to contribute to the risk of developing asthma at a population level. Recent reports have identified three further genes by positional cloning approaches.13 Even larger populations will be needed to study epigenetic and gene–environment interactions effectively in order to derive an accurate predictive model for the risk of an individual developing asthma.
Note added in proof
Since submitting this paper, a further family based association study of ADAM33 polymorphisms has been published, confirming the lack of marked association with single SNPs in ADAM33 with asthma in a moderate sized sample.14
Acknowledgments
The authors thank Esteban Burchard and Ngim Ung for providing data from the GALA study.
REFERENCES
Footnotes
-
Sources of support: none
-
Conflict of interest: none
Linked Articles
- Airwaves