Article Text

Abstract
Background: Chronic inflammation and airway remodelling are characteristics of chronic obstructive pulmonary disease (COPD). Hyaluronan (HA) is an extracellular matrix compound with proinflammatory activity. HA levels in induced sputum from patients with COPD were measured and related to local inflammation. The expression of hyaluronan synthase 2 (HAS2) and hyaluronidase 2 (HYAL2) was analysed in lung tissue.
Methods: Sputum was obtained from 18 patients with COPD (forced expiratory volume in 1 second (FEV1) 62% predicted (range 20–76)) and 14 healthy smokers. HA and inflammatory markers were measured using ELISA assays. Lung sections were obtained from five patients with severe COPD (FEV1 <30%) and from five smokers, and mRNA levels of HAS2 and HYAL2 were analysed by polymerase chain reaction.
Results: HA levels were significantly higher in the sputum from patients with COPD than controls. The COPD population appeared to consist of two subpopulations with either high or moderate HA levels. The subgroup of patients with high HA levels had lower FEV1 than the moderate HA group. In addition, neutrophil influx and levels of interleukin-8, and the soluble tumour necrosis factor receptors R55 and R75 were significantly higher in patients with high HA levels than in those with moderate HA levels and controls. Semiquantitative analysis revealed enhanced expression of HYAL2 in lung tissue of patients with severe COPD compared with control subjects.
Conclusion: These data indicate a relationship between HA levels, local inflammation and severity of disease, and suggest enhanced breakdown of HA in the lungs of patients with COPD.
- COPD, chronic obstructive pulmonary disease
- ECM, extracellular matrix
- FEV1, forced expiratory volume in 1 second
- HA, hyaluronan
- IL, interleukin
- TNF, tumour necrosis factor
- chronic obstructive pulmonary disease
- hyaluronan
- inflammation
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- ECM, extracellular matrix
- FEV1, forced expiratory volume in 1 second
- HA, hyaluronan
- IL, interleukin
- TNF, tumour necrosis factor
Chronic obstructive pulmonary disease (COPD) is a clinical syndrome varying from chronic obstructive bronchiolitis to emphysema, and represents a major cause of morbidity and mortality. Airway inflammation is a key feature of COPD and is thought to have a role in its pathogenesis.1 There is a marked increase in neutrophils and inflammatory mediators such as interleukin (IL)-8 and soluble tumour necrosis factor receptor P55 (sTNFR55) in bronchoalveolar lavage (BAL) fluid and sputum from patients with COPD, which correlates with disease severity.2,3 Bronchial biopsy of patients with mild disease show infiltration of macrophages and CD8+ T cells which is related to airflow limitation whereas, in patients with severe disease, influx of neutrophils and macrophages is reported to be associated with severity of obstruction.4,5
Exposure to environmental contaminants—of which cigarette smoke is the most evident—is a known risk factor for the development of COPD. However, smoking cessation does not appear to result in resolution of inflammation although improvement of respiratory symptoms is observed.6,7 These studies indicate that there are mechanisms that maintain the chronic inflammatory process in patients with COPD once it has been established.
Chronic pulmonary inflammation and lung injury are associated with damage, repair and remodelling of the extracellular matrix (ECM). One ECM compound of special interest is hyaluronan (HA), which is present in increased amounts in the serum and BAL fluid of patients with various inflammatory diseases.8,9 HA is an ubiquitously distributed component of the ECM that exists in healthy tissues as a high molecular weight non-sulfated glycosaminoglycan polymer composed of repeating disaccharide units of glucuronic acid and N-acetylglucosamine. In the ECM, HA plays a role in water homeostasis, plasma protein distribution and matrix structure.10 Increasing evidence indicates additional biological functions of HA which seem to be dependent on its molecular size. Oligosaccharides of HA have been shown to induce angiogenesis and endothelial cell proliferation,11 and also to activate dendritic cells.12 Low molecular weight HA fragments, with an average molecular mass of 250 kDa, stimulate production of cytokines, chemokines, and matrix metalloproteinase 12 by macrophages.13,14 Similar activating effects have been shown with eosinophils15 and renal epithelial cells.16 Furthermore, high molecular weight HA has been found to have protective effects for osteoarthritis and rheumatoid arthritis.17
To study whether HA is involved in the pathogenesis of COPD, HA levels in the sputum of patients with COPD were analysed and related to local inflammation and severity of disease. In addition, lung resection material was used to analyse the expression of two HA modulating enzymes—hyaluronan synthase 2 (HAS2) and hyaluronidase 2 (HYAL2).
METHODS
Subjects
Induced sputum was obtained from 18 patients with smoking related COPD prospectively recruited from general practice. COPD was defined by GOLD criteria, indicating a stable airflow limitation with post bronchodilator forced expiratory volume in 1 second (FEV1) of <80% predicted for age and height, in combination with a ratio of FEV1 to forced vital capacity (FVC) of <70.18 A history of respiratory diseases other than COPD as well as increased respiratory complaints or respiratory tract infection during the 4 weeks preceding the study were criteria for exclusion. None of the patients used inhaled steroids, 13 patients were prescribed combination therapy (fenoterol/ipratropium bromide) on a regular basis, two patients were on long acting β2 agonists, and all other patients were prescribed bronchodilating agents on demand. A control group of 14 so-called “healthy smokers” was recruited with a normal FEV1 and no medical history of lung disease.
Lung specimens were obtained from five smokers with severe COPD (FEV1 <30%) undergoing lung volume reduction surgery for emphysema and from five smokers with no airflow obstruction who underwent lung resection for a solitary peripheral tumour with no medical history of chronic lung disease. The control subjects lived in the same geographical area as the patient population. The study was approved by the medical ethics committee of the University Hospital of Maastricht. Written informed consent was obtained from all subjects.
Pulmonary function testing
FEV1 was measured by trained lung function technicians using a spirometer (Masterlab; Jaeger, Würzburg, Germany) before and 15 minutes after inhalation of β agonist via a metered dose inhaler.
Sputum induction, processing, and analysis of inflammatory mediators
Sputum was induced using the method developed by Keatings2 as reported previously.3 IL-8, sTNFR55 and sTNR75 were measured using a sandwich ELISA as described elsewhere.19,20 Inter-batch and intra-batch variability of the IL-8, sTNFR55 and sTNFR75 ELISA was <10%. Dithiothreitol (DTT, 0.1%), which was used for sputum processing, did not affect measurement of these parameters.3
Analysis of HA in sputum
HA was measured using an enzyme linked absorbent assay as described elsewhere21 with some modifications. Immuno-Maxisorp plates (Nunc, Roskilde, Denmark) were coated overnight at room temperature with 2.5 μg/ml HA (Sigma-Aldrich, St Louis, MO, USA) dissolved in phosphate buffered saline (PBS). Plates were washed in PBS 0.05% (w/v) Tween 20 (PBS/tween) and then blocked for 1 hour in 1% (w/v) BSA in PBS/tween. Samples (60 μl) or standard HA in the range 1–2000 ng/ml were added to a 96-well sample plate (Greiner GmbH, Frinckenhause, Germany) and, after addition of an equal amount of 0.7 μg/ml biotin labelled HA binding protein (Seikagaku, Tokyo, Japan), this mixture was immediately administered to the Immuno-Maxisorp plates. After incubation for 1 hour the plates were washed and subsequently incubated with streptavidin-peroxidase conjugate (Zymed Laboratories Inc, San Fransisco, CA, USA). 3,3′,5,5′-tetramethylbenzidine (KPL, Gaithersburg, MD, USA) was used as the substrate and absorbance was measured spectrophotometrically at 450 nm using a micro ELISA autoreader.
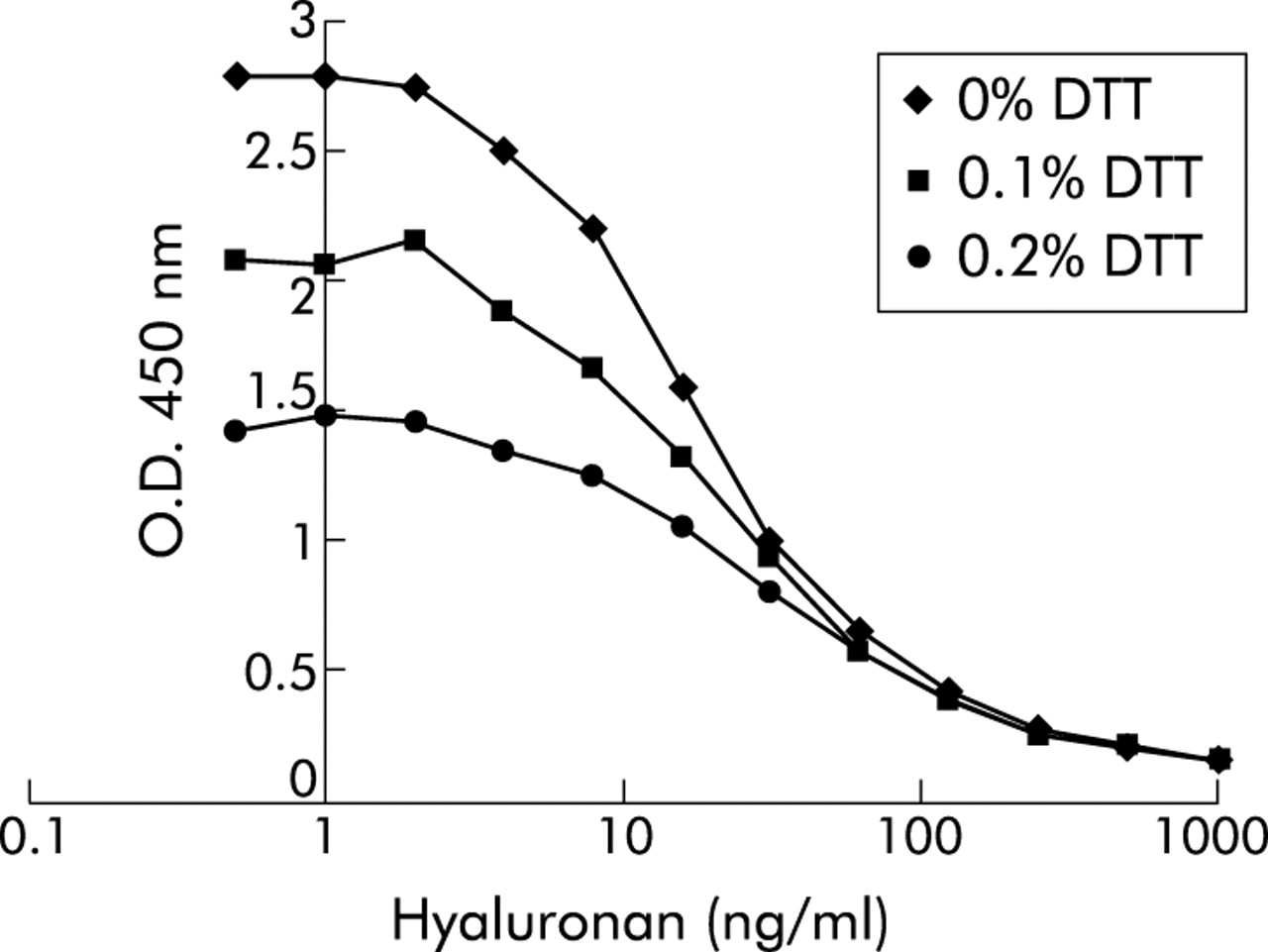
Since sputum samples were processed using 0.1% DTT, the effect of DTT on the HA assay was analysed. Addition of DTT to standard HA was found to suppress the assay concentration dose dependently, but the maximal optical density of standard curves was still sufficiently high to be useful (fig 1). In addition, the competition curves had slopes which paralleled the standards without DTT. The assay was therefore performed by pre-incubating HA standards with 0.1% DTT for 20 minutes (similar to the processing of the sputum samples3) before adding the HABP-biotin. To check for the presence of HA binding proteins in sputum which could interfere with the measurement of HA, six samples were treated with papain (Sigma-Aldrich) as described previously.21 This resulted in a recovery rate of 95% (range 63–143) compared with the untreated samples. Furthermore, spiking of 100 ng/ml HA in sputum (n = 16) gave a recovery rate of 125% (range 56–177) after correction for endogenous HA, indicating that no interfering factors were present in the sputum.
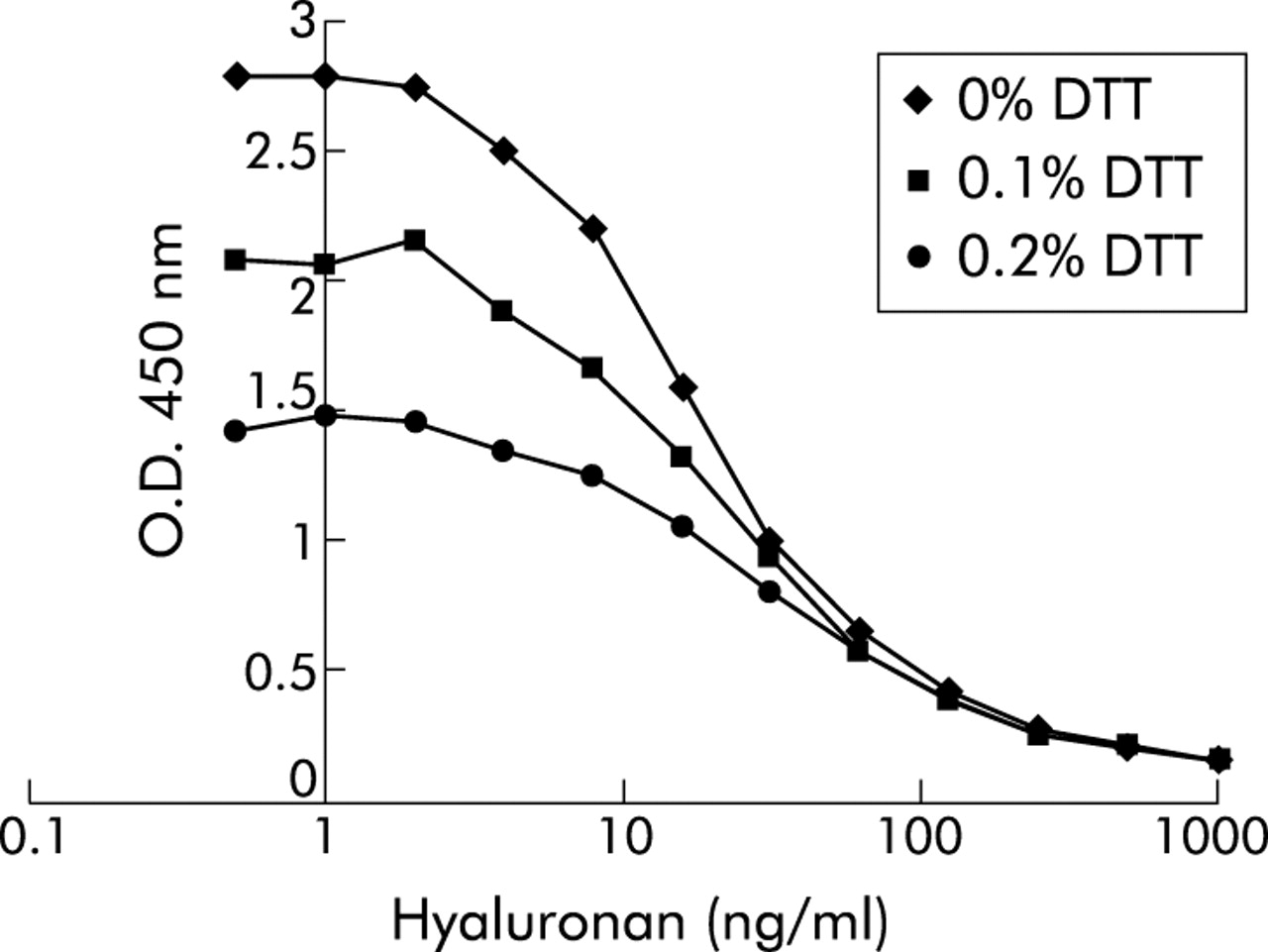
Effect of dithiothreitol (DTT) on recovery of hyaluronan (HA) standards in immunosorbent assay. Plates were coated overnight with HA, washed in PBS 0.05% (w/v) Tween 20 (PBS/tween) and then blocked for 1 hour in 1% (w/v) BSA in PBS/tween. HA standards were prepared in PBS/0.05% tween buffer containing 0%, 0.1% or 0.2% DTT and incubated for 20 minutes at room temperature in a 96-well sample plate. After addition of an equal volume of biotin labelled HA binding protein, this mixture was immediately applied to the HA coated plates. After incubation for 1 hour the plates were washed and incubated with streptavidin-peroxidase conjugate. Detection was with 3,3′,5,5′-tetramethylbenzidine substrate and absorbance was measured spectrophotometrically at 450 nm. One representative experiment out of a series of three is shown.
RNA isolation and RT-PCR
Total RNA of homogenised human lung tissue was obtained using the RNAqueous total RNA isolation kit from Ambion (St Austin, Texas, USA), treated with DNAse, and cDNA was then obtained by reverse transcription of RNA. For standardisation of the different RNA samples, polymerase chain reaction (PCR) of two housekeeping genes (glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin) was employed on serial dilutions of cDNA. PCR was performed in 25 μl reaction volume containing 100 μM of each dNTP, 200 nM primers, and 0.5 U Taq DNA polymerase (Perkin Elmer/Cetus, Emeryville, CA, USA). Human PCRs were performed as described in table 1. The PCR products were separated by electrophoresis in ethidium bromide stained 1.2% agarose gel and visualised by UV illumination (Imagemasters VDS, Uppsala, Sweden).
RT-PCR conditions
Statistical analysis
The results are presented as median (range). Groups (n = 3) were compared using the Kruskal-Wallis H test; in cases where there were statistical differences (p<0.05), further analysis of subsets of groups (n = 2) were performed using the Mann-Whitney U test or the Fisher’s exact test (to compare categorical variables). SPSS version 10.0 for Windows (SPSS Inc, Chicago, IL, USA) was used.
RESULTS
Induced sputum was collected from 18 patients with COPD and 14 control subjects. Patients with COPD had significant airflow limitation (FEV1 % predicted 62 (range 20–76)) whereas control subjects had normal FEV1 values, as shown in table 2. The mean age of the patients (89% of whom were men) was 61 years (range 41–74). Twelve of the 18 patients were still smoking and the mean pack-years of the whole patient group was 40 (range 21–122).
Clinical characteristics of controls and subgroups of COPD patients with moderate and high HA levels
Analysis of HA levels in the soluble phase of the sputum showed the presence of this ECM compound in all samples. Moreover, HA levels were significantly higher in COPD patients than in control subjects (median 234 ng/ml (range 64–1000) v 66 ng/ml (range 34–423); p = 0.001, fig 2). The patients with COPD appeared to consist of two subpopulations with either high (n = 6) or moderate (n = 12) HA levels (1000 (912–1000) ng/ml v 201 (64–374) ng/ml). Based on this observation, post hoc analysis was performed in which the two subgroups were compared with each other and with controls. A comparison of the subgroup of patients with moderate HA levels with control subjects still revealed a significant difference (p<0.05).

Individual hyaluronan (HA) levels in induced sputum from 18 patients with chronic obstructive pulmonary disease (COPD) and 14 healthy smokers. Median values are indicated by the horizontal bars. The Mann-Whitney U test was used to test for differences between groups. *p<0.005 v healthy controls.
The two subgroups of COPD patients were further characterised according to their clinical characteristics (table 2). The subgroup with high HA levels tended to have lower FEV1 levels than those with moderate HA levels (p = 0.061). Furthermore, this subgroup consisted mainly of ex-smokers while the subgroup with moderate HA levels consisted mainly of current smokers. This relationship between smoking status and HA levels was not present in the control subjects (HA levels 52 ng/ml (34–423) in current smokers and 79 ng/ml (53–239) in ex-smokers; p = 0.2).
To analyse the relationship between HA levels and local inflammation, the subgroups of COPD patients were compared with healthy smokers with respect to inflammatory cells and markers. There were significantly increased numbers of cells in the sputum of patients with high HA levels than in patients with moderate HA levels or control subjects (table 3). This increase in cell numbers was caused primarily by increased numbers of neutrophils in the sputum, while no differences were seen in the numbers of macrophages, eosinophils or lymphocytes. Analysis of the levels of the inflammatory markers IL-8 and the soluble forms of the TNF receptors R55 and R75 showed that, as for cell numbers, these mediators were only increased in the subgroup of patients with high HA levels (fig 3). No differences were observed between the subgroup of patients with moderate HA levels and control subjects.
Total and differential cell counts in sputum of control subjects and subgroups of COPD patients with moderate and high levels of HA

Levels of interleukin (IL)-8, soluble tumour necrosis factor receptor 55 (sTNFR55) and sTNFR75 in induced sputum of healthy smokers and subgroups of patients with chronic obstructive pulmonary disease (COPD) with moderate hyaluronan (HA) and high HA levels. The Kruskal-Wallis H test was used to test for differences between groups (n = 3, p<0.05) and the Mann-Whitney U test was used to test for differences between subgroups (n = 2). *p<0.005 between subgroups.
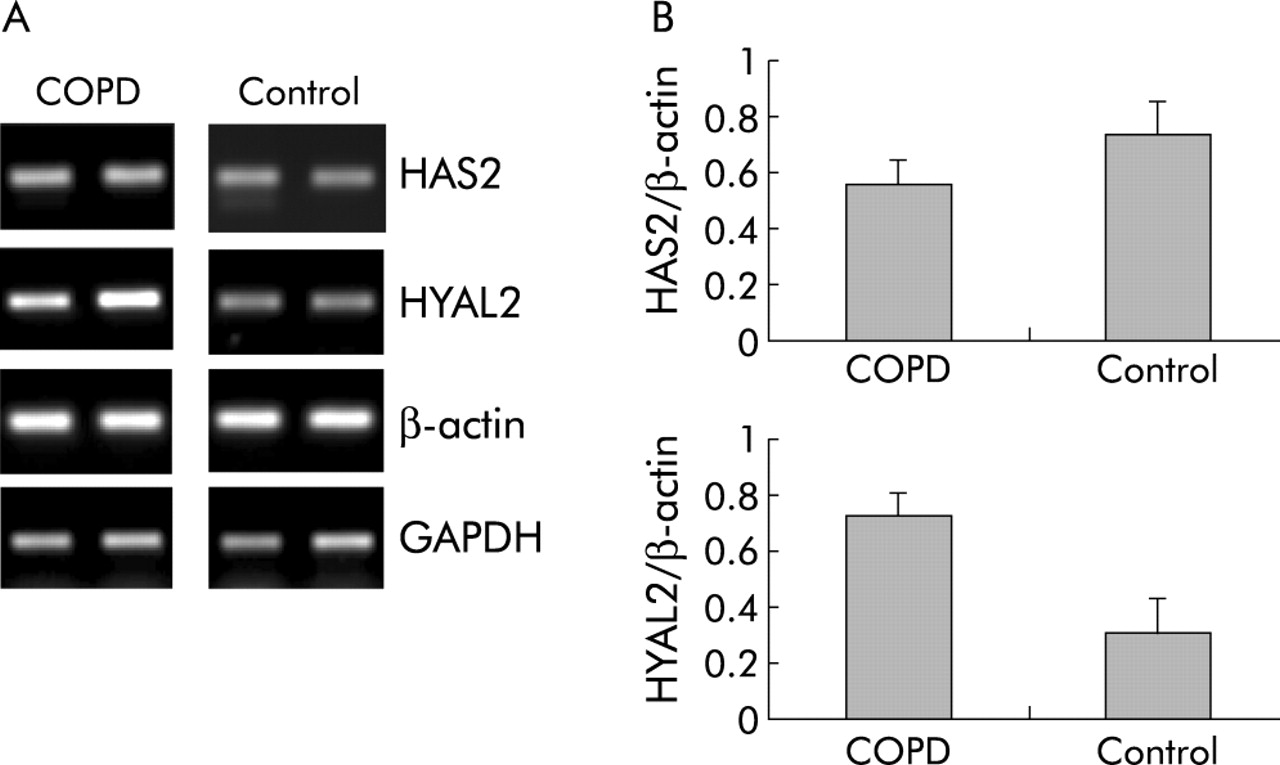
HA is known to have a rapid turnover rate so the mRNA expression of HAS2, which is involved in the synthesis of HA, and of HYAL2, which degrades the HA compound, was investigated. Lung tissue from five patients with COPD who underwent lung volume reduction surgery and five smoking controls was analysed. The clinical features are shown in table 4. No difference was seen in the expression of HAS2 between the two groups (fig 4) but the expression of HYAL2 was higher in patients with COPD than in control subjects, suggesting enhanced degradation of HA in COPD.
Clinical characteristics of subjects who underwent resection
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HAS2 and HYAL2 expression in lung tissue from patients with COPD and control subjects determined by RT-PCR. (A) mRNA expression of HAS2 (314 bp), HYAL2 (330 bp), and the house keeping genes GAPDH (300 bp) and β-actin (294 bp) of two representative subjects from both study groups. The equal intensity of the GAPDH and β-actin mRNA bands confirmed equal amounts of cDNA in RT-PCR. (B) Densitometric ratios of HAS2/β-actin and HYAL2/β-actin in patients with COPD (n = 5) and control subjects (n = 5). Data are mean (SE).
DISCUSSION
There is increasing evidence to indicate that integrity and balance of ECM components, which continuously undergo degradation and reconstruction, are essential for normal lung function and the response to injury. COPD is characterised by a slowly progressive irreversible airflow obstruction due to loss of lung elasticity resulting from parenchymal destruction and peripheral airflow limitation.1 Remodelling of the ECM in COPD is considered to be caused by an ongoing local inflammatory process. However, changes in the composition of the ECM could also contribute to inflammation, as indicated in recent studies which have shown the proinflammatory activity of the ECM compound HA.11–16 Further proof of the importance of HA in inflammation is evident from animal models showing decreased inflammation with the use of HA binding peptides to sequester HA.22,23 Moreover, recent evidence indicates that HA based cables, formed by stressed cells, are able to bind CD44 containing inflammatory cells and, in this way, to engage leucocytes recruited to the tissue by an inflammatory stimulus.24
In this study we have shown, for the first time, increased levels of HA in the sputum of patients with COPD. Moreover, a subgroup of patients with high HA levels was defined with a tendency towards more severe airflow limitation than patients with moderate HA levels, indicating a relationship between HA and disease severity. This subgroup of patients with high HA levels consisted mainly of ex-smokers (five of six), confirming other studies which have shown that the inflammatory process persists after smoking cessation.6,7 In line with our results, increased HA levels were also found in BAL fluid from patients with COPD.25,26 Moreover, an inverse relationship has been observed between BAL fluid HA levels and lung function measurements in young patients with COPD,25 comparable to our data.
Activated neutrophils are considered to have a central role in chronic airway inflammation in COPD.1,3,5 Results from the present study showed a marked sputum neutrophilia and higher levels of the soluble markers of inflammation (IL-8, sTNFR55 and sTNFR75) in the subgroup of patients with high HA levels than in those with moderate HA levels and controls, thus emphasising the relationship between HA and local inflammation. The relatively high neutrophil count seen in smoking controls in this study compared with other studies2 may be due to the older age of these subjects, which has been shown to be positively related to the percentage of polymorphonuclear leucocytes.27 No enhancement of these inflammatory markers was observed in the subgroup of COPD patients with moderate HA levels, suggesting that HA is a more sensitive marker for COPD than the other inflammatory markers studied to date.
Under physiological conditions HA in the lung is cleared by alveolar macrophages and, in addition, it is mobilised to the lymphatic system where it is taken up via receptor mediated pathways.28 It is possible that one or both pathways might be disturbed in COPD, thus contributing to enhanced HA levels. In line with this hypothesis, reduced binding and clearance of HA by alveolar macrophages has been demonstrated in models of fibrosis.29 Alternatively, since bacteria such as group A streptococcus are known to form polysaccharide capsules composed of HA,30 they cannot be excluded as a possible source of HA. However, no relationship was found between bacterial load and HA levels in bronchial BAL fluid of patients with chronic bronchitis.26 Further studies are needed to elucidate the origin of HA in sputum.
To obtain an insight into the metabolism of HA in COPD, the expression of HAS2 and HYAL2—which have been shown to be involved in HA turnover during the early phase of lung injury31—were studied. This study shows, for the first time, enhanced expression of HYAL2, but not of HAS2, in the lungs of patients with severe COPD compared with control subjects. This is in line with observations in the bleomycin induced alveolitis model in which enhanced HA levels were paralleled by increased hyaluronidase activity.32 HYAL2 is a membrane associated enzyme which was reported to degrade HA into fragments of 20 kDa,33 suggesting that small HA fragments are formed in COPD lungs. In addition, the observation that reactive oxygen species can degrade HA34 and the presence of an oxidant/antioxidant imbalance in COPD1,35 further suggest the presence of depolymerised HA. Since the biological activity of HA is determined by its molecular size, studies are currently in progress to determine the size of HA in the lungs of patients with COPD.
The increased expression of HYAL2 suggests enforced breakdown of HA from connective tissue in the lungs of patients with COPD which is supported by the observation of reduced HA levels in the lungs of patients with emphysema.36 As studies by Cantor et al37 indicate that HA protects elastic fibres against disruption by proteases, HA degradation may also result in an exaggerated breakdown of the ECM. Whether enhanced expression of HYAL2—now shown in the peripheral tissue—is also present in the central airways of patients from which induced sputum is derived38 still needs to be explored.
Cellular activation by low molecular weight HA was found to be mediated by the principal cellular HA receptor CD44.13,15 CD44 is constitutively expressed on leucocytes and parenchymal cells including endothelium, epithelial, and smooth muscle. Although most CD44 positive cells fail to exhibit CD44 receptor function, HA binding to CD44 can be enhanced under proinflammatory conditions, which is mediated by the action of cytokines like TNF.39,40 Indeed, enhanced expression of CD44 has been demonstrated in several inflammatory diseases including asthma.41 Further studies need to be performed on CD44 expression and HA binding ability of cells in patients with COPD.
In summary, enhanced levels of the ECM compound HA were found in the sputum of patients with mild to severe COPD. The presence of HA seems to be related to disease severity and to markers of local inflammation. HA was shown to be a sensitive marker for COPD, with increased levels seen in a group of patients with mild COPD in which the inflammatory markers IL-8, sTNFR55 and sTNFR75 were not enhanced. In addition, increased expression of HYAL2 was observed in the lungs of patients with severe COPD, suggesting enhanced breakdown of HA. Further studies need to be performed to elucidate the origin of this HA and its role in the chronic inflammation seen in COPD.
Acknowledgments
The authors thank Dr A M W J Schols for expert assistance with the statistical evaluation of the data, M P van Spijk and Greetje Castelijn for technical assistance concerning the PCR experiments, and dr J van den Born (Department of Molecular Cell Biology, VU University Medical Centre, Amsterdam) for the protocol for HA measurement.
REFERENCES
Footnotes
-
This study was supported by a grant from the Netherlands Asthma Foundation.