Article Text

Abstract
Background: Most patients with cystic fibrosis (CF) have a ΔF508 mutation resulting in abnormal retention of mutant gene protein (ΔF508-CFTR) within the cell. This study was undertaken to investigate ΔF508-CFTR trafficking in native cells from patients with CF with the aim of discovering pharmacological agents that can move ΔF508-CFTR to its correct location in the apical cell membrane.
Method: Nasal epithelial cells were obtained by brushing from individuals with CF. CFTR location was determined using immunofluorescence and confocal imaging in untreated cells and cells treated with sildenafil. The effect of sildenafil treatment on CFTR chloride transport function was measured in CF15 cells using an iodide efflux assay.
Results: In most untreated CF cells ΔF508-CFTR was mislocalised within the cell at a site close to the nucleus. Exposure of cells to sildenafil (2 hours at 37°C) resulted in recruitment of ΔF508-CFTR to the apical membrane and the appearance of chloride transport activity. Sildenafil also increased ΔF508-CFTR trafficking in cells from individuals with CF with a single copy ΔF508 (ΔF508/4016ins) or with a newly described CF trafficking mutation (R1283M).
Conclusions: The findings provide proof of principle for sildenafil as a ΔF508-CFTR trafficking drug and give encouragement for future testing of sildenafil and related PDE5 inhibitors in patients with CF.
- cystic fibrosis
- sildenafil (Viagra)
- genetics
Statistics from Altmetric.com
Cystic fibrosis (CF) is caused by mutations in the CF gene protein, the cystic fibrosis transmembrane conductance regulator (CFTR).1 By far the most common mutation is ΔF508, present on 70–80% of chromosomes worldwide. Wild type CFTR resides at the apical membrane of epithelial cells where it acts as a cyclic AMP-dependent Cl− channel1 and regulates other ion channels2 and protein secretion.3 Defective trafficking of ΔF508-CFTR leads to abnormal retention of mutant protein within the cell and consequent loss of CFTR function.1 The reported degree of mislocalisation of ΔF508-CFTR in native epithelial cells varies between different tissues and types of preparation.4–7 More recently we and others have developed a technique to study the location of CFTR in freshly isolated native airway epithelial cells from patients with CF which allows quantitation of the percentage of cells with a defined CFTR location.8,9 We have shown a marked mislocalisation of ΔF508-CFTR which was corrected by the CFTR activating benzo[c]quinolizinium compounds.8 Thus, although our studies and those of others suggest that at least some ΔF508-CFTR traffics to the apical membrane in native CF cells,7–9 the finding that the majority of ΔF508-CFTR is mislocalised8 emphasises that moving ΔF508-CFTR to the apical membrane is necessary for developing a drug treatment targeted at CFTR rescue.
Arising from our earlier studies showing correction of antibody inhibited CFTR function by cyclic nucleotide phosphodiesterase (PDE) inhibitors,10 we now show that the PDE5 inhibitor sildenafil (Viagra) increases ΔF508-CFTR trafficking. Since this is a drug in clinical use that causes rapid movement of ΔF508-CFTR from within the cell to the apical membrane, the results represent a major step forward in the development of a treatment that addresses the basic cellular abnormality for the majority of patients with CF.
METHODS
Patients and cell sampling
Airway epithelial cells were obtained by nasal brushing from three non-CF individuals (age 9–22 years) and from six CF individuals (age 6 months to 16 years) undergoing flexible bronchoscopy under sedation as previously described.8 The CF individuals had the following genotypes: four ΔF508/ΔF508, one ΔF508/4016ins, and one R1283M/E60X. The study was approved by the local ethics committee of Bro Taf Health Authority.
CFTR localisation in nasal epithelial cells
Immediately following nasal brushing, the brushes were placed in DMEM/F12 medium and the attached cells incubated in the presence or absence of sildenafil (150 μM) for 2 hours at 37°C. Cells were then fixed and CFTR localised by immunofluorescence as previously described.8 Briefly, after incubation, cells were smeared gently onto “Snowcoat X-tra” microslides (Surgipath), left to air dry, fixed for 5 minutes at −20°C in 5% acetic acid in ethanol, washed in phosphate buffered saline (PBS) containing (in mM): 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, 1.4 KH2PO4, pH 7.4 and stored at −20°C. Following treatment with blocking reagent (normal rabbit serum 1:20), cells were incubated with primary antibody overnight at 4°C, washed three times, and incubated with FITC conjugated secondary antibody (1:100) for 30 minutes at room temperature. The CFTR antibody used was our well characterised anti-CFTR antibody MPCT1 (affinity purified C-terminal; 1:100).8 Slides were then mounted in Fluorosave Reagent (Calbiochem, La Jolla, CA, USA) and fluorescence was detected using confocal laser scanning microscopy on a Leitz DMRBE microscope (Leica, Germany) fitted with a TCS4D scanner (Leica, Germany).
Measurement of chloride transport activity in CF15 cells
The SV40 transformed nasal epithelial cell line JME/CF15 from a patient with CF (ΔF508/ΔF508)11 was cultured in DMEM/F12 supplemented with adenine (180 µM), insulin (5 µg/ml), transferrin (5 µg/ml), hydrocortisone (1.1 µM), tri-iodothyronine (2 nM), epinephrine (5.5 µM), epidermal growth factor (1.64 nM) and 10% FCS. Chloride channel activity was assayed by measuring the rate of iodide (125I) efflux.8,12 CF15 cells were incubated for 2 hours at 37°C in the presence or absence of either sildenafil or MPB-91 (250 μM). Cells were then washed with efflux buffer containing (in mM): 137 NaCl, 4.4 KCl, 0.3 KH2PO4, 0.3 NaH2PO4, 4.2 NaHCO3, 1.3 CaCl2, 0.5 MgCl2, 0.4 MgSO4, 5.6 glucose and 10 HEPES, pH 7.5 and incubated in efflux buffer containing 1 µM KI and 1 µCi Na125I /ml (NEN, Boston, MA, USA) for 2 hours at 37°C. Cells were then washed with efflux buffer. After 1 minute the buffer was removed and quickly replaced by 300 µl of the same buffer. The first four aliquots were used to establish a stable baseline in efflux buffer alone. Efflux buffer containing forskolin (10 µM) and genistein (30 µM) to stimulate CFTR activity was used for the remaining aliquots. At the end of the incubation the buffer was recovered and cells solubilised in 1 ml 1 N NaOH. The radioactivity was determined using a gamma counter (Cobra II, Packard Bell).
Analysis of data
For CFTR localisation up to 140 cells for each condition were examined, categorised as having a distinct CFTR location of either near nucleus or apical, and expressed as a percentage of all cells counted. Only ciliated tall columnar epithelial cells were counted as these can be readily differentiated from non-ciliated columnar epithelial cells and basal cells, even when ΔF508-CFTR is localised close to the nucleus.
For CFTR chloride transport activity the fraction of initial intracellular 125I lost during each time point was determined and time dependent rates (k) of 125I efflux were calculated from ln (125It1/125It2)/ (t1 − t2) where 125It is the intracellular 125I at time t, and t1 and t2 are successive time points.8,12 Curves were constructed by plotting k versus time. Relative rates were calculated and correspond to kpeak − kbasal (per minute). Concentration-response curves were constructed by plotting the percentage activation as a function of the concentration of sildenafil (100% corresponds to the maximum relative rate obtained at the highest concentration, i.e. 1.5 mM). Differences between treated and untreated cells were examined using the Student’s t test and p values of <0.05 were considered significant.
RESULTS
Mislocalisation of ΔF508-CFTR in CF nasal cells
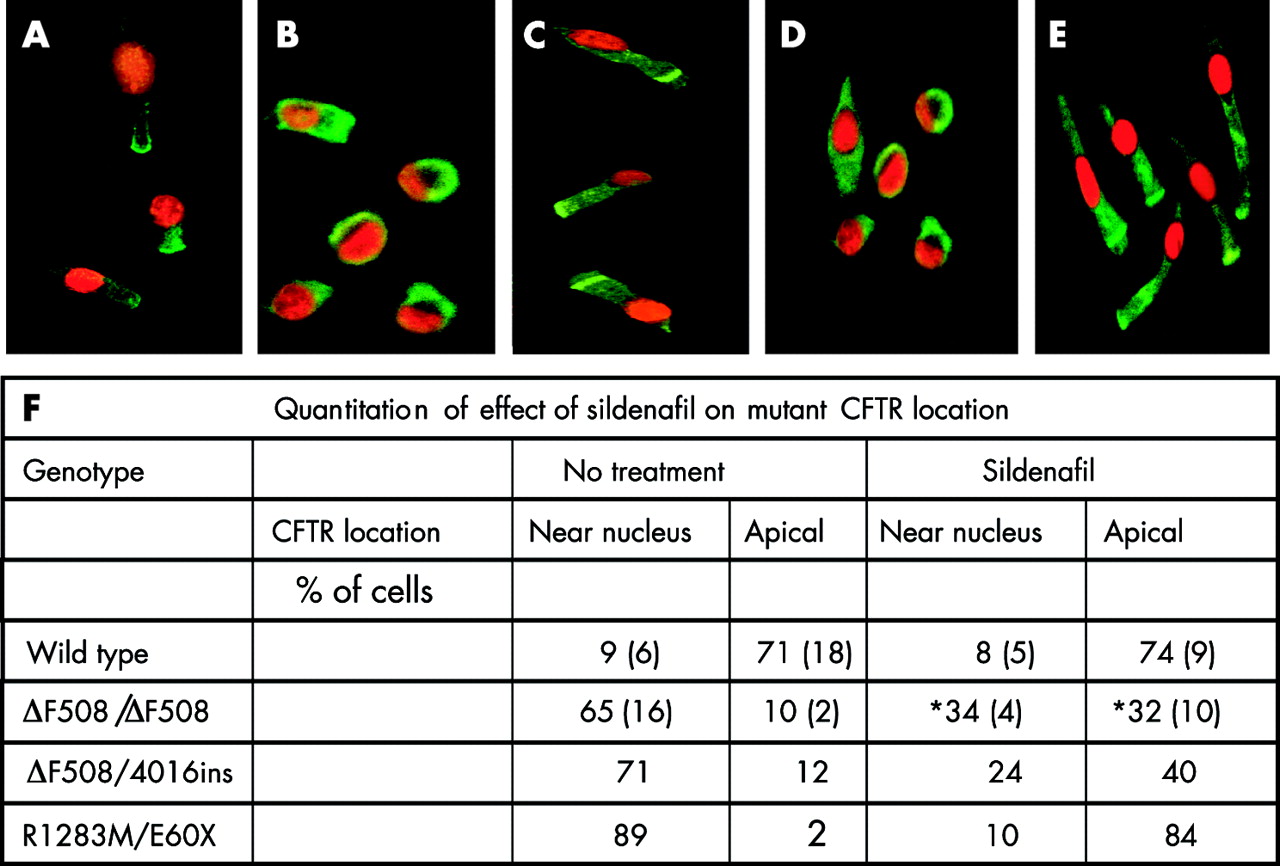
As in our previous studies,8 nasal epithelial cells obtained by brushing were polarised with cilia visible at the apical end of the cell that continued to beat for the 2 hour incubation before fixation. The nasal cells expressed cytokeratins typical of epithelial cells throughout the cell. In cells from non-CF individuals, wild type CFTR was located at the apical end of the cell (fig 1A). In untreated cells from ΔF508/ΔF508 CF individuals and from a CF individual with the genotype ΔF508/4016ins, 60–70% of cells showed a near nuclear location of ΔF508-CFTR with only 10% having ΔF508-CFTR present at the apical membrane (fig 1B and F). This is in agreement with our previous data on the location of CFTR in wild type, ΔF508/ΔF508 and ΔF508/G551D cells.8

Immunofluorescent images of non-CF and CF nasal epithelial cells showing that sildenafil corrects abnormal mutant CFTR location in CF cells. Cells were incubated for 2 hours at 37°C without sildenafil (A, B, D) or with 150 μM sildenafil (C, E). Images show CFTR immunofluorescence in green with the nucleus counterstained with propidium iodide in red. The results are representative of cells from (A) three non-CF individuals, (B, C) four ΔF508/ΔF508 CF individuals, and (D, E) one R1283M/E60X individual. They show that both ΔF508- and R1283M- CFTR are predominantly restricted within the cell to a distinct location adjacent to the nucleus (near nucleus) in untreated cells (B, D) whereas, after sildenafil treatment, many cells have an apical location of CFTR (C, E). (F) Cells were examined (up to 140 cells for each condition) and categorised as having a distinct CFTR location of either near nucleus or apical. The remainder of both untreated and treated cells showed a more even distribution of CFTR throughout the cell. Data are mean (SD). *p<0.05 for difference from no treatment.
Actions of sildenafil on ΔF508-CFTR location
In ΔF508/ΔF508 cells treated with sildenafil (150 µM for 2 h at 37°C), a marked change in ΔF508-CFTR location was observed (fig 1C and F) with a significant decrease (p<0.05) in the percentage of cells having a near nuclear location and a significant increase (from 10% to 32%, p<0.05) in cells having ΔF508-CFTR at the apical membrane. CFTR was also relocated in cells with a single copy ΔF508 genotype (ΔF508/4016ins) following sildenafil treatment (fig 1F). The data indicate that CFTR trafficking drugs such as sildenafil will be potentially useful in all CF patients carrying a ΔF508 mutation. Although the concentration of sildenafil effective in increasing ΔF508-CFTR trafficking is higher than might be achieved in plasma (1 μM) following a single oral dose (100 mg) of Viagra (data from Pfizer: www.Viagra®.com), nevertheless the data show proof of principle for investigating this relatively safe class of compounds for use in CF. Furthermore, the effect of sildenafil was selective in that the distinct locations of endoplasmic reticulum (p115), Golgi (GM130), and 20S proteasome proteins were not altered with sildenafil treatment (data not shown) and sildenafil (150 µM, 2 hours) had no effect on wild type CFTR location (fig 1A, F) in cells from non-CF individuals.
A new CF trafficking mutation
Cells from a CF individual heterozygous for a rare missense mutation, R1283M,13 and a stop mutation, E60X, were also examined (fig 1D–F). As for ΔF508 expressing cells, the results (fig 1D, F) showed severe mislocalisation of CFTR with the majority of cells having mutant CFTR within the cell at a site close to the nucleus (near nuclear location). The defective location of this mutant CFTR was also markedly corrected by sildenafil (fig 1E, F) with the majority of treated cells showing an apical CFTR location. This is the first description of R1283M as a trafficking mutation and sildenafil was effective in correcting the defect. The results indicate that trafficking mutations in both the first and second nucleotide binding domains (NBD1 and NBD2) of CFTR are targets for sildenafil.
Effect of sildenafil on CFTR chloride transport activity in CF15 cells
The human airway epithelial CF cell line CF15 was used as a model to study CFTR dependent chloride transport.11 In untreated CF15 cells there was no stimulation of 125I efflux in response to the CFTR agonists forskolin and genistein (fig 2A) consistent with its ΔF508/ΔF508 CF origin.11 However, when cells were treated for 2 hours at 37°C with sildenafil a significant stimulation by forskolin and genistein was recovered (fig 2A and B). The effect of sildenafil was similar to that of the potent trafficking drug MPB-91,8 although of lower magnitude. The concentration dependence of the effect of sildenafil treatment showed an EC50 of 718 (1) μM (fig 2C, D). Thus, CF15 cells were less sensitive to the effect of sildenafil, requiring a higher concentration than native cells from CF patients to restore function. The effects of a range of Cl− channel blockers on the forskolin/genistein stimulated iodide efflux of sildenafil treated cells (1 mM for 2 hours at 37°C) was determined. The efflux was inhibited by glibenclamide and DPC but not by DIDS or calixarene, demonstrating that sildenafil had recruited functional ΔF508-CFTR Cl− channels at the apical membrane. The results indicate that, like MPB-91, sildenafil was able to restore the chloride channel activity of ΔF508-CFTR at 37°C. Immunolocalisation of ΔF508-CFTR in CF15 cells indicated that, following treatment for 2 hours at 37°C with 1 mM sildenafil, ΔF508-CFTR had moved from within the cell to an apical location (data not shown).

{kind=link}
{kind=link}
Correction of the chloride channel activity of CFTR in CF15 human airway epithelial cells by sildenafil. Cells were incubated for 2 hours at 37°C with or without drug and then CFTR Cl− channel activity measured by iodide efflux as described in the Methods section. All data in (A)–(F) are mean (SE) for four separate experiments. (A), (C) and (E) show representative traces from single assays. The presence of forskolin (10 µM) and genistein (30 µM) is indicated by the horizontal bar. (B), (D) and (F) show mean (SE) data for each experimental condition as indicated. (A) □, no drug; ▴, sildenafil (1 mM); ▪, MPB-91 (250 μM). (B) ***p<0.001 for difference from untreated cells; NS, no significant difference. (C) and (D) Concentration dependence of the effect of sildenafil treatment (2 hours at 37°C). (C) ▪, 300 μM; ▴, 500 μM; ▾, 1 mM; ⧫, 1.1 mM; •, 1.3 mM; □, 1.5 mM sildenafil. (D) Concentration-response curve showing % activation at each sildenafil concentration. (E) and (F) Effect of chloride channel inhibitors on iodide efflux stimulated by forskolin (10 µM) and genistein (30 µM) from cells treated with sildenafil (1 mM) for 2 hours at 37°C. (E) □, basal; ▪, no blocker; ▴, +100 nM calixarene; ▾, + 500 μM DIDS; ⧫, + 100 μM glibenclamide; •, + 500 μM DPC. (F) Histograms showing the relative rate (kpeak − kbasal) for the experimental conditions indicated below each bar. ***p<0.001 for difference from cells stimulated by forskolin and genistein (fsk+gst) without the inhibitor; NS, no significant difference.
DISCUSSION
This study has shown, for the first time, a dramatic and rapid effect of the PDE5 inhibitor sildenafil in directing ΔF508-CFTR to the apical membrane in native CF airway cells from patients with CF. The effect is comparable to our previously demonstrated effect of MPB compounds.8 By comparison, other non-selective treatments such as low temperature (25–27°C), glycerol, or 4-phenylbutyrate have required long term exposure to show modest effects on ΔF508-CFTR trafficking.14 The technique that we have developed for confocal immunofluorescence localisation of wild type and ΔF508-CFTR in native nasal epithelial cells from control and CF individuals allows CFTR location to be visualised in individual cells, the pattern of mislocalisation quantified, and the actions of pharmacological agents on ΔF508-CFTR trafficking evaluated. In agreement with our previous studies8 and those of others,7,9 ΔF508-CFTR was predominantly mislocalised and present within the cell. ΔF508-CFTR was detectable at the apical membrane in a minority (approximately 10%) of cells.
The present studies have also identified a new CFTR trafficking mutation in NBD2 (R1283M) that has not previously been investigated. The only other report of altered trafficking of CFTR with a mutation in NBD2 is N1303K.15 The present results, showing that sildenafil corrects ΔF508- and R1283M-CFTR location, both mutations in nucleotide binding domains, will provide insights into the mechanism of action of drugs that increase CFTR trafficking. It is noteworthy that neither sildenafil nor MPB compounds affect the trafficking of wild type CFTR, nor of other normal ER or Golgi proteins, thus providing a selective action. We have previously provided evidence16 that the speed and selectivity of the effect of MPB compounds on ΔF508-CFTR trafficking is due to direct binding to the CFTR protein at a site within the first cytoplasmic domain. It is therefore possible that sildenafil also binds directly to ΔF508-CFTR, although an action through cyclic GMP signalling has not been ruled out.
There is agreement that ΔF508-CFTR has substantial Cl− channel activity when present at the apical membrane, which some studies suggest is comparable to that of wild-type CFTR.1 Thus, the present study showing recruitment of ΔF508-CFTR to the apical membrane in native CF cells in response to the drug sildenafil, is a major step forward in restoring CFTR function. Although the concentration of sildenafil shown to traffic ΔF508-CFTR in a CF cell line was higher than in native cells, sensitivities may vary between cell lines, isolated native cells, and in vivo, and this will form the basis of future studies. Nevertheless, the finding that sildenafil, a relatively safe drug already in clinical use, has a dramatic effect in increasing ΔF508-CFTR trafficking will assist in the discovery of new derivatives and is a major advance towards developing a rational drug treatment aimed at repair or rescue of activity of the mutant CF gene protein. The present results indicate that an approach to drug therapy using CFTR trafficking drugs is likely to be applicable to the majority of patients with CF.
REFERENCES
Footnotes
-
Financial support was provided by WORD, Pfizer Pharmaceuticals Group, Vaincre la Mucoviscidose (VLM), the Cystic Fibrosis Trust, UK and the Laurence Goodchild fellowship.
-
Conflicts of interest: none.
Linked Articles
- airwaves