Article Text

Abstract
Background: Auto-antibodies against granulocyte-macrophage colony stimulating factor (GM-CSF) may be central to the pathogenesis of adult sporadic pulmonary alveolar proteinosis (PAP). The role of anti-GM-CSF auto-antibodies in paediatric forms of PAP is as yet unclear.
Methods: Anti-GM-CSF auto-antibodies were determined with the help of an antigen capture assay using serum and/or bronchoalveolar lavage (BAL) fluid from 27 patients with PAP (nine adults, 15 children, three neonates) and from 185 children with different diseases as disease controls (various pulmonary conditions and patients with malignancies).
Results: Anti-GM-CSF auto-antibodies were detected in the serum of five of seven adult PAP patients. They were not found in the serum of any of the children or neonates with PAP nor in any of the disease control patients. Raised anti-GM-CSF titres were found in BAL fluid from three of four adult patients with PAP. Anti-GM-CSF auto-antibodies were detected in BAL fluid of only one of the 15 children (age at diagnosis 11 years, age at BAL 24 years) and in none of the neonates with PAP, nor in any of the disease control patients.
Conclusions: The presence of anti-GM-CSF auto-antibodies seems to define an autoimmune disease underlying most of the adult sporadic type of PAP, but age at diagnosis may cause an overlap with children in some rare instances. In most of the children and all of the neonates the anti-GM-CSF titres were not significantly increased, indicating that alternative explanations are needed for the pathogenesis of the disease in these patients.
- ABCA3, human gene encoding ATP binding cassette transporter A3
- BAL, bronchoalveolar lavage
- CAP, congenital alveolar proteinosis
- CD36, human gene encoding leucocyte differentiation antigen (scavenger receptor CD36)
- CF, cystic fibrosis
- CSF2, human gene encoding colony stimulating factor 2 (GM-CSF)
- CSF2RA, human gene encoding GM-CSF receptor α subunit
- CSF2RB, human gene encoding GM-CSF receptor β subunit (common to interleukins 3 and 5)
- Csfgm, murine gene encoding GM-CSF
- GM-CSF, granulocyte-macrophage colony stimulating factor
- LPI, lysinuric protein intolerance
- OLB, open lung biopsy
- PAP, pulmonary alveolar proteinosis
- PAS, Periodic acid Schiff
- PPAR-γ, human gene encoding peroxisome proliferator-activated receptor gamma
- rh-GM-CSF, recombinant human GM-CSF
- SFTPB, human gene encoding SP-B
- SFTPC, human gene encoding SP-C
- SP-B, surfactant associated protein B
- SP-C, surfactant associated protein C
- TBB, transbronchial biopsy
- children
- pulmonary alveolar proteinosis
- granulocyte-macrophage colony stimulating factor (GM-CSF)
- auto-antibodies
Statistics from Altmetric.com
- ABCA3, human gene encoding ATP binding cassette transporter A3
- BAL, bronchoalveolar lavage
- CAP, congenital alveolar proteinosis
- CD36, human gene encoding leucocyte differentiation antigen (scavenger receptor CD36)
- CF, cystic fibrosis
- CSF2, human gene encoding colony stimulating factor 2 (GM-CSF)
- CSF2RA, human gene encoding GM-CSF receptor α subunit
- CSF2RB, human gene encoding GM-CSF receptor β subunit (common to interleukins 3 and 5)
- Csfgm, murine gene encoding GM-CSF
- GM-CSF, granulocyte-macrophage colony stimulating factor
- LPI, lysinuric protein intolerance
- OLB, open lung biopsy
- PAP, pulmonary alveolar proteinosis
- PAS, Periodic acid Schiff
- PPAR-γ, human gene encoding peroxisome proliferator-activated receptor gamma
- rh-GM-CSF, recombinant human GM-CSF
- SFTPB, human gene encoding SP-B
- SFTPC, human gene encoding SP-C
- SP-B, surfactant associated protein B
- SP-C, surfactant associated protein C
- TBB, transbronchial biopsy
- children
- pulmonary alveolar proteinosis
- granulocyte-macrophage colony stimulating factor (GM-CSF)
- auto-antibodies
Pulmonary alveolar proteinosis (PAP) is a rare respiratory disease characterised by the accumulation of surfactant derived material in the lung of patients.1 New insights into the pathogenesis of the disease have had a great impact on the nosological classification. Today, PAP can be categorised into three different classes: congenital PAP, acquired PAP, and secondary PAP.2,3
The congenital form of PAP is characterised by an acute onset immediately after birth with respiratory distress and rapid progression.4 An undefined proportion of congenital cases are caused by different mutations of the gene encoding SP-B (SFTPB, MIM 178640), with a frameshift indel (g.1549C>GAA, or “121ins2”) being the predominant but not exclusive genetic lesion leading to complete SP-B deficiency.5,6 Another possible cause of congenital PAP might be a mutation in the gene encoding the GM-CSF receptor β subunit (CSF2RB, MIM 138981).7 As lung transplantation has been the only known treatment for the congenital forms of PAP so far and not all the congenital cases can be explained by SP-B deficiency or a GM-CSF receptor defect, new studies of the pathogenesis are warranted with a view to developing alternative therapeutic strategies.
The acquired form of PAP is characterised clinically by cough, dyspnoea, and progression to respiratory failure, with repetitive whole lung lavage being the current standard treatment.1,3 As GM-CSF knock-out mice develop an interstitial and storage lung disease reminiscent of the human PAP,8,9 animal studies have been carried out and have shown a correction of this alveolar proteinosis phenotype in Csfgm-/- mice10 by inhalation of aerosolised recombinant human GM-CSF (rh-GM-CSF).11 These animal studies and the detection of auto-antibodies against GM-CSF in the serum and bronchoalveolar lavage (BAL) fluid from patients with acquired PAP have resulted in a better understanding of the pathogenesis of the human disease.12 The presence of anti-GM-CSF auto-antibodies in serum is not only of diagnostic value in adult patients,13,14 but rh-GM-CSF administered to adults with PAP—either subcutaneously or by inhalation—has resulted in a significant benefit to single patients15–17 and in prospective studies. Seymour et al18 were able to show a clear improvement in the clinical symptoms in five out of 14 patients while, in another study, six out of 11 patients responded to rh-GM-CSF therapy.19 Although the latter authors postulated that the anti-GM-CSF antibody titre is predictive of the response to treatment,19 the exact role of anti-GM-CSF auto-antibodies in the pathogenesis of adult PAP has not been fully elucidated.20
There is a Gaussian distribution of acquired PAP with respect to age at onset, although there is a slight over-representation of cases below the age of 10 years.3 However, none of the published studies linking GM-CSF to PAP has included juvenile cases. Five infants with congenital PAP in the absence of an SFTPB mutation have already been treated with rh-GM-CSF without a clinical or haematopoietic response,21 and without any prior evidence for a pathogenic relationship between the presence of anti-GM-CSF auto-antibodies and PAP in the congenital or juvenile cases. There is therefore a need to clarify the role of anti-GM-CSF auto-antibodies in patients with PAP from different age groups.
METHODS
Subjects
Controls, disease controls and comparison groups
The presence of anti-GM-CSF auto-antibodies was assessed in the serum and/or BAL fluid from 12 healthy adults and 185 children with various lung diseases including atopy, oncological haematology, general paediatric pneumonology, tracheostoma, and cystic fibrosis (CF). All subjects were in a clinically stable state when sampled. None of the subjects in this group had clinical or radiological evidence of PAP and none had received rh-GM-CSF. The characteristics of the different groups of subjects are shown in table 1. All materials from controls, disease controls, and comparison groups were anonymised.
Characteristics of subject groups
Patients with alveolar proteinosis
Twenty seven patients were diagnosed with PAP, nine of whom were adults, 15 children and three neonates. The diagnosis was made from the respiratory symptoms with a special focus on the clinical course and radiographic and chest CT findings, and the diagnosis was confirmed by open or transbronchial lung biopsy. In all patients the characteristic histological pattern of alveolar filling with PAS positive material was present. The disease was in an active state in all patients. There were no cases of secondary PAP due to Pneumocystis jiroveci infection or to other known external causes.3 Lysinuric protein intolerance (LPI, MIM 222700) had been ruled out as a rare underlying syndromic cause of PAP.22 Further clinical data of the children and neonates with PAP are shown in tables 2 and 3.
Details of disease onset in children and neonates with PAP
Clinical details of the children with PAP
The study was approved by the local institutional review boards and informed consent was obtained from all patients or their legal custodian(s).
Adults
Because the presence of anti-GM-CSF antibodies in serum has been used as a hallmark of adult patients with sporadic PAP, nine such patients were included in the study, five of whom were treated at the Hospitals for Pulmonary Disease, Gauting and Grafschaft in the Federal Republic of Germany and a further four have been described elsewhere.23
Children
Twelve of the 15 children with PAP have recently been reported in detail with a specific focus on SP-B and its encoding gene.6 Three further children with PAP, confirmed by lung histology, had been attending our hospital on a regular basis. All these patients had dyspnoea and chronic cough, and most of them showed failure to thrive and required repetitive therapeutic bronchoalveolar lavage.6 Three of the 15 children died from severe respiratory distress. A detailed description of the children with PAP is given in tables 2 and 3, the URD codes referring to the paper by Tredano et al.6
Neonates
All three neonates with PAP have also been reported in detail with a special focus on SP-B and its encoding gene.6 Two of them had typical BAL findings indicative of congenital alveolar proteinosis (CAP), and the diagnosis in the third neonate of intra-alveolar storage disease similar to PAP was confirmed by open lung biopsy.6SFTPB mutation was not found in any of the cases, and locus exclusion could be obtained in two of them by family segregation analyses. All patients died from severe respiratory distress. Further details are given in table 2. As it is still arguable whether complete SP-B deficiency should be considered as PAP or rather regarded as a disease entity in its own right, neonates with complete SP-B deficiency were not included in this study, especially when found to be mutated at SFTPB.
Bronchoalveolar lavage (BAL) fluid and serum samples
Immediately after collection of the lavage fluid (4×1 ml/kg 0.9% NaCl instilled via a bronchoscope or 1–2 ml/kg via an end hole catheter in intubated neonates) and serum samples, the cells were removed by centrifugation (200g, 10 minutes) and the supernatants were stored at −70°C until assayed.
Antigen capture assay for the detection of anti-GM-CSF auto-antibodies
All measurements were performed in a central laboratory (Lung Research Group, Children’s Hospital of Ludwig Maximilians University, Munich, Germany). The assay was performed as described by Kitamura et al.12,13 Briefly, serum samples were diluted 750 and 1500 fold with Tris buffered saline (TBS) containing 0.1% Triton X-100 (Sigma Chemicals, St Louis, MO, USA) and BAL samples were used undiluted and in a 1:4 dilution. 50 μl of diluted or undiluted samples (the TBS/TC buffer alone being used as a control) were transferred onto plates coated with 1 μg/ml rh-GM-CSF (gift of Novartis Pharma AG, Basel, Switzerland) for 1 hour at 37°C and blocked with 1% bovine serum albumin in TBS/TC for 1 hour at 37°C. After incubation for 1 hour at 37°C, the plates were washed three times with TBS/TC and the antibodies captured by rh-GM-CSF were detected by peroxidase labelled rabbit anti-human IgG antibody (Dako Corporation, Carpinteria, CA, USA). After washing three times, colour was developed using ABTS (Roche Diagnostics, Mannheim, Germany) and absorbance was measured at 405 nm (Biolise Software, anthos htIII). The serum samples from the adult patients with PAP were further diluted 6000, 12 000, and 18 000 fold and the BAL fluid samples from these patients were further diluted 10, 50, and 100 fold. Serum and BAL fluid samples from adult PAP patients with known GM-CSF antibodies served as positive controls in each assay.
Statistical analysis
The results of the tests were expressed as fold of the optical density (OD) of the blank value run in each assay. Median OD readings on the blank for the serum assays were 0.09 (interquartile range (IQR) 0.08–0.1) (range 0.07–0.14), and median OD readings on the blank for the BAL assays were 0.1 (IQR 0.08–0.14) (range 0.07–0.3). Statistical differences between two groups were analysed by a two tailed Wilcoxon test, a p value of ⩽0.05 being considered significant.
RESULTS
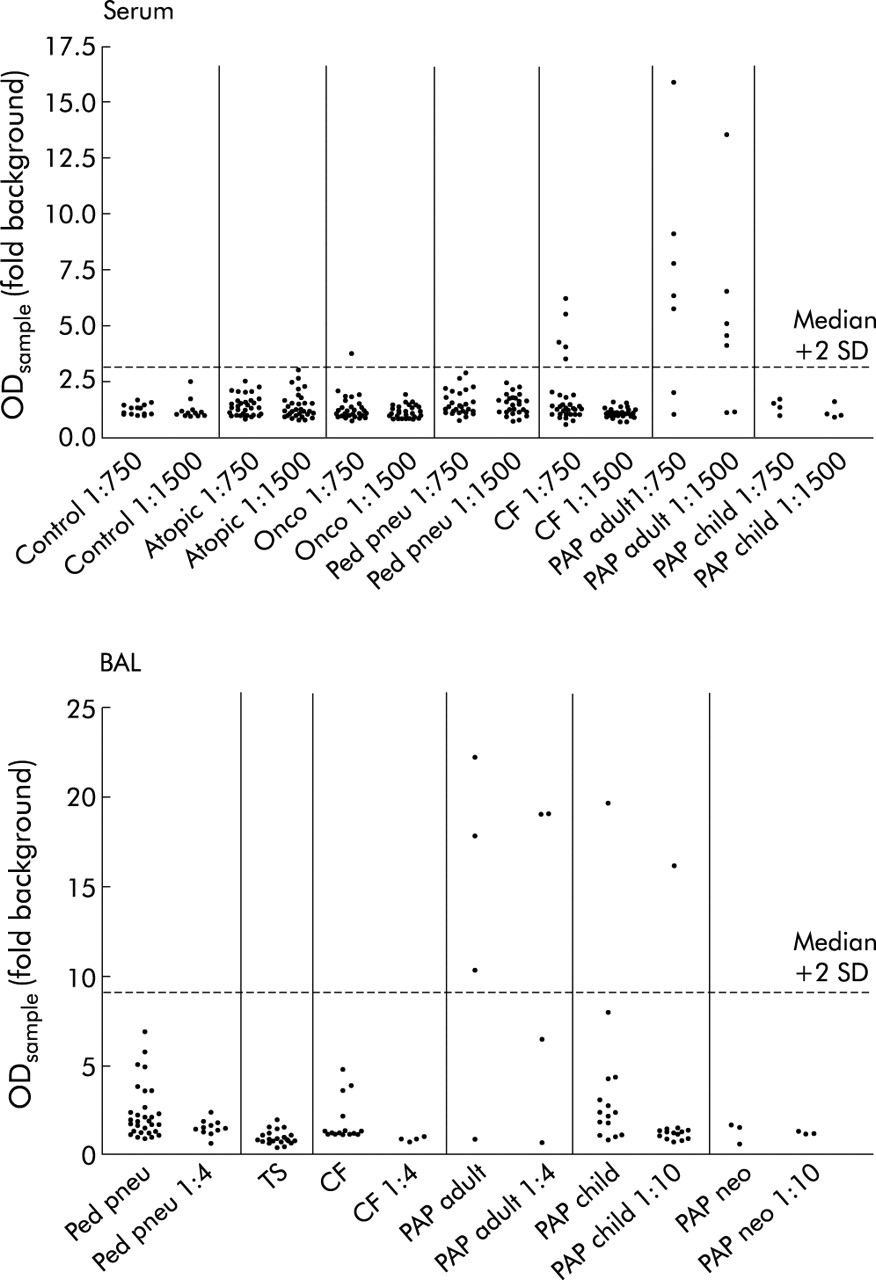
All antibody levels are expressed as fold background level. In serum samples anti-GM-CSF auto-antibody levels among the controls, the disease controls, and the comparison groups did not differ and were on average 1.2 (IQR 1.0–1.5) (range 0.6–6.2), n = 145 at 1:750 fold dilution and 1.1 (IQR 1.0–1.4) (range 0.7–3.0), n = 145 at 1:1500 fold dilution. At 1:750 dilution one out of 38 oncological patients and five of 33 CF patients had increased titres which disappeared after dilution to 1:1500 (fig 1A). Among the patients with PAP, five of seven patients with the adult form had increased levels of anti-GM-CSF auto-antibody at titres that were clearly detected at 1:1500 dilution. Further dilution of these serum samples resulted in a concentration dependent reduction in the antibody level (fig 2A). In contrast, in none of the children with PAP were significant anti-GM-CSF auto-antibody titres detected (fig 1A).

Anti-GM-CSF auto-antibody titres determined by antigen capture assay in (A) serum and (B) bronchoalveolar lavage (BAL) fluid from controls, various disease comparison groups, and patients of all age groups diagnosed with PAP. Serum samples were diluted 1:750 and those with an OD greater than 1.2 fold of background were also assessed at 1:1500 fold dilution. BAL samples were used undiluted and those with an OD above the 75th percentile further diluted 1:4 or 1:10 fold. Data points from individual patients and duplicate determinations are given and expressed as multifold of the OD of the blank sample as run in parallel in each assay.

{kind=link}
{kind=link}
Dilution of anti-GM-CSF auto-antibody titre expressed as multifold of the OD of blank samples in (A) serum samples (seven adult patients with PAP) and (B) bronchoalveolar lavage (BAL) fluid (four adult patients with PAP).
In BAL fluid anti-GM-CSF auto-antibody levels in disease controls and comparison groups did not differ from each other and were on average 1.2 (IQR 0.8–1.9) (range 0.4–6.8), n = 69 without prior dilution. The samples in which anti-GM-CSF auto-antibody levels were increased above the 75th percentile were further diluted and those levels were on average 1.3 (IQR 0.9–1.6) (range 0.5–2.3), n = 15 at 1:4 fold dilution. Anti-GM-CSF auto-antibody levels were found to be significantly increased in one of 15 BAL fluid samples from children with PAP and in none of the BAL fluid samples from neonates with PAP (fig 1B). However, three out of four patients with adult PAP had high auto-antibody titres that decreased in proportion to dilution (fig 2B).
DISCUSSION
In agreement with previous findings,12,14 we found anti-GM-CSF auto-antibodies in all but two adult patients with PAP. With the exception of one patient whose disease onset was at 11 years of age—but BAL fluid was obtained at the age of 24 years—none of the children and none of the neonates with PAP had raised levels of anti-GM-CSF auto-antibodies. These increased antibody titres are deemed specific because they were absent in the paediatric disease groups whose conditions included atopy, oncological haematology, general paediatric pneumonology, tracheostoma, and CF, and were also absent in the healthy controls.
Adult PAP
Bonfield et al14 postulated that anti-GM-CSF auto-antibodies were highly specific for adult idiopathic PAP. Our findings are consistent with this postulate, but in our study not all adult PAP patients would fit into this pathophysiological group. Indeed, based on the two adult patients without raised antibodies in this study, we found that the sensitivity of anti-GM-CSF autoantibody determinations in adult PAP is not complete. The details of one of these patients has been published elsewhere as one of a series of four patients.23 This patient differed from the other three in that she had normal GM-CSF release after stimulation of peripheral blood monocytes with lipopolysaccharide (LPS) while the others had a diminished response to LPS. She also had normal GM-CSF levels in cells harvested by BAL. The second patient in whom anti-GM-CSF auto-antibodies were not raised in the serum and BAL fluid was diagnosed at the age of 35 years, has been followed for the last 6 years, and has undergone five therapeutic whole lung lavages. Her diagnosis was confirmed by HRCT scanning, macroscopic and microscopic examination of BAL fluid, and transbronchial biopsy findings. Because all known causes for secondary PAP have been ruled out, hitherto unknown aetiologies for adult PAP could be present in these two patients.
Juvenile PAP
In all but one patient with juvenile PAP no increase in the level of anti-GM-CSF auto-antibodies was found in either the serum or BAL fluid. These patients were devoid of an SFTPB mutation,6 and other known external or syndromic causes of PAP such as LPI had been ruled out. A GM-CSF receptor defect was assessed and excluded in four of the 15 patients from whom specific samples were made available.24 These findings differ from those of earlier publications with a focus on adult sporadic PAP and suggest that alternative explanations should be sought in these patients. The risk of recurrence of juvenile PAP in siblings5 indicates that the underlying pathogenetic defect might be at least partly inherited. Consistent with this view, an endogamous population with a high prevalence of juvenile PAP has been reported.25 An exception to this hypothesis is provided by case 14 (table 2) in whom high titres of anti-GM-CSF auto-antibodies were found in BAL fluid samples in a reproducible manner. As previously reported, no SFTPB or SFTPC mutation was found in this patient. Unfortunately no serum has been available so far and BAL fluid was available from the age of 24 years only. It is still uncertain therefore whether this patient should be categorised as juvenile or adult PAP. Although we lack information on the course of antibody levels from childhood through adolescence, we have assumed that this patient is a case of acquired PAP with disease onset in childhood and have categorised him as a juvenile form of PAP.
Congenital PAP
No anti-GM-CSF auto-antibodies were found in any of the three patients with congenital PAP. These patients were devoid of an SFTPB6 or SFTPC26 mutation and, akin to juvenile PAP patients, other known external or syndromic causes of PAP such as LPI had been ruled out. One of the cases (no 16) was a consanguineous patient with an affected sibling (the two other cases were non-consanguineous sporadic cases). Thus, although no mutation has been found in these neonatal patients, it is likely that an endogenous genetic aetiology is involved. The clinical course was particularly severe in these patients and all died due to severe respiratory distress. There is therefore clinical overlap with complete SP-B deficiency, but the presence of SP-B in BAL fluid and/or immunohistochemistry (with absent SFTPB mutation) indicates a distinct entity. As with juvenile PAP, congenital PAP might therefore represent a group with at least one entity in its own right which differs from both complete SP-B deficiency and acquired adult PAP with GM-CSF antibodies. In children with congenital PAP disease onset occurs directly after birth. In contrast, children with the juvenile form of PAP do not show symptoms after birth but start developing them months or even years later (table 3). Therefore, although the pathophysiology might be comparable, from a clinical point of view the juvenile form of PAP might be categorised as “acquired PAP” and can be differentiated from “congenital PAP”.
Children with various lung diseases
In order to assess the specificity of anti-GM-CSF auto-antibodies throughout childhood, we measured anti-GM-CSF auto-antibodies in BAL fluid and serum samples from children with various lung diseases. Anti-GM-CSF auto-antibodies were not found in any of these 185 subjects nor in the healthy adult controls, using a cut off point of the median +2SD. It should be noted, however, that low anti-GM-CSF auto-antibody titres were detected in some of these patients (five CF and one oncology patient at a 1:750 dilution, fig 1). These findings are important as the exact specificity of anti-GM-CSF auto-antibodies is not as yet entirely clear. In 1999 Kitamura et al12 did not find anti-GM-CSF auto-antibodies in BAL fluid from 53 healthy adult controls and 14 adult disease controls or in serum samples from 30 healthy adults. However, the same group reported increased levels of anti-GM-CSF auto-antibodies in serum samples from two of 40 healthy adults while these antibodies were absent from BAL fluid from 40 disease controls.13 From these findings and those published by Bonfield et al14 (who detected slightly increased levels of anti-GM-CSF auto-antibodies in serum from two out of 23 disease and 21 healthy adult controls), it can be suggested that significantly increased anti-GM-CSF auto-antibodies are not likely in non-PAP patients or healthy controls. The occurrence of anti-GM-CSF auto-antibodies in serum or BAL fluid makes the diagnosis of acquired PAP very likely.
GM-CSF treatment in PAP
While whole lung lavage remains the gold standard for treating adult PAP patients,3 a significant clinical improvement has been shown in about half of adult PAP patients treated with rh-GM-CSF.18,19 Rh-GM-CSF therapy has also been attempted in congenital PAP (without SP-B deficiency) but with no visible benefit. All such patients had a normal haematopoietic response to rh-GM-CSF.21 Thus, there is no pathophysiological rationale to date for the use of rh-GM-CSF in early onset PAP, a form that is not likely to be acquired and not likely to be related to a defect of the GM-CSF pathway. Furthermore, it has even been shown that, following administration, immunocompetent patients treated with rh-GM-CSF may develop antibodies capable of binding GM-CSF.27,28 Further studies of the pathogenesis of congenital and juvenile PAP are urgently needed before more patients with these early onset forms of PAP are treated with rh-GM-CSF although screening negative for GM-CSF auto-antibodies.
Conclusion
Less than one third of cases with unexplained neonatal respiratory distress have a combination of SFTPB gene mutations entailing SP-B deficiency,6 whereas others may harbour a mutation in the gene encoding for ATP binding cassette transporter A3 (ABCA3, MIM 267450).29 An unknown proportion of affected neonates and infants could harbour a mutation in CSF2RB, the gene encoding the GM-CSF receptor β subunit,7 while a sizeable proportion of neonates or children with unexplained respiratory distress (including PAP) are likely to harbour a genetic defect disrupting any of the processes involved in surfactant homeostasis which has not yet been identified.
Raised levels of anti-GM-CSF auto-antibodies suggesting disruption of the GM-CSF signalling pathway as an important clue in the pathogenesis of acquired PAP can be seen in most forms of acquired PAP. Whether these GM-CSF auto-antibodies occur only in adult patients or start developing in childhood is uncertain. As several patients with acquired PAP (mostly children) do not have GM-CSF antibodies, analysis of genes whose products are related to this pathway such as CSF2 (MIM 138960), CSF2RA (MIM 306250), CSF2RB, PPAR-γ (MIM 601487) or CD36 (MIM 173510)30 could help to unravel some of the additional causes for the disease pathogenesis in these juvenile forms of acquired PAP.
REFERENCES
Footnotes
-
Supported by a grant from DFG Gr 970/7-1.