Article Text

Abstract
Compared with the effects of chronic smoke exposure on lung function and airway inflammation, there are few data on the acute effects of smoking. A review of the literature identified 123 studies investigating the acute effects of cigarette smoking on inflammation and oxidative stress in human, animal, and in vitro models. An acute smoking model is a relatively easy and sensitive method of investigating the specific effects of cigarette smoke on oxidative stress and inflammation. Acute smoke exposure can result in tissue damage, as suggested by increased products of lipid peroxidation and degradation products of extracellular matrix proteins. Acute cigarette smoke has a suppressive effect on the number of eosinophils and several inflammatory cytokines, possibly due to the anti-inflammatory effect of carbon monoxide. An acute smoking model can supplement other ways of studying the effects of smoking and is an as yet underinvestigated method for intervention studies in smoking related diseases.
- ACS, acute cigarette smoking
- AMs, alveolar macrophages
- BALF, bronchoalveolar lavage fluid
- CO, carbon monoxide
- COPD, chronic obstructive pulmonary disease
- CS, cigarette smoke
- CSE, cigarette smoke extract
- EIC, elastase inhibitory capacity
- GSH, reduced glutathione
- GSSG, oxidised glutathione
- HO-1, heme oxygenase-1
- IFN-γ, interferon-γ
- IL, interleukin
- NE, neutrophil elastase
- NO, nitric oxide
- PMNs, polymorphonuclear cells
- TBARS, thiobarbituric acid reactive substances
- TEAC, trolox equivalent antioxidant capacity
- TNF-α, tumour necrosis factor α
- chronic obstructive pulmonary disease
- smoking
- inflammation
- oxidative stress
Statistics from Altmetric.com
- ACS, acute cigarette smoking
- AMs, alveolar macrophages
- BALF, bronchoalveolar lavage fluid
- CO, carbon monoxide
- COPD, chronic obstructive pulmonary disease
- CS, cigarette smoke
- CSE, cigarette smoke extract
- EIC, elastase inhibitory capacity
- GSH, reduced glutathione
- GSSG, oxidised glutathione
- HO-1, heme oxygenase-1
- IFN-γ, interferon-γ
- IL, interleukin
- NE, neutrophil elastase
- NO, nitric oxide
- PMNs, polymorphonuclear cells
- TBARS, thiobarbituric acid reactive substances
- TEAC, trolox equivalent antioxidant capacity
- TNF-α, tumour necrosis factor α
Chronic obstructive pulmonary disease (COPD) is a worldwide leading cause of morbidity and mortality and its prevalence is still rising.1 It is therefore important to understand the development of this disease in order to develop strategies of prevention, treatment, and cure. In the past decade research has focused on the pathophysiological mechanisms underlying the development of COPD, yet several questions remain unanswered.
Most studies investigating the role of smoking in the pathophysiology of COPD have been carried out in chronic smokers. The drawback of studying the effects of actual smoke exposure in persistent smokers is the likely effect of already developed structural changes in the airways on the response to smoke. It is therefore important to study the response to the first smoke exposure of a “naïve” lung in order to assess the relevant changes that may have a role in the first steps of COPD development. In addition, an acute smoking model could be attractive for future intervention studies. We hypothesise that an acute smoking model can give clear and more specific information about the pathophysiological mechanisms of smoking induced lung disease.
In this paper we review the literature on the acute effects of smoking. We focus on human, animal, and in vitro models and systematically describe the effects of acute smoke exposure on the cellular response, specifically on oxidative stress and inflammatory mediators. We also review similarities and discrepancies in the smoking response between the three model systems and discuss how these results relate to the current insights on the development of COPD.
METHODS
The Medline, OldMedline, Winspirs and Cochrane Library databases were searched from their inception until October 2003. The language used was limited to English. Firstly, a database including all articles on the effects of smoking on pulmonary status was composed (keywords “cigarette smoke, tobacco smoke” and all subheadings and “lungs, pulmonary” and all subheadings). Secondly, a selection was made of the articles describing the acute effects of smoking (keyword “acute”). Thirdly, all articles describing the acute effects of smoking on oxidative stress, inflammatory mediators, and inflammatory cells in humans, animals, and in vitro models were selected. Fourthly, a specific search was done on oxidative stress (keywords “oxidative stress” and all subheadings). Acute smoking was defined as an effect measured during the 24 hours after smoke exposure. It is explicitly mentioned when articles on chronic smoking or COPD have been used. Only studies describing mainstream cigarette smoke were included, the number of cigarettes smoked not being a selection criterion.
RESULTS
Acute effects of cigarette smoke in humans
Twenty five studies examining the acute effects of cigarette smoking (ACS) in humans were identified (see table S1 available online at www.thoraxjnl.com/supplemental), 16 on inflammation and nine on oxidative stress.
All studies were performed in chronic smokers with normal lung function. In 13 studies smokers were instructed to refrain from smoking before the acute smoke exposure, varying between 7 and 24 hours. Ten studies did not provide information on this and in two studies the subjects were not instructed to refrain from smoking.
Inflammatory cells
In chronic smoking the numbers of neutrophils are increased in the blood and bronchoalveolar lavage fluid (BALF).2–4 With ACS both increased5 and unchanged numbers of neutrophils have been reported in BALF.6 Acute smoke exposure had no effect on the number of monocytes or the total number of leucocytes in BALF.6 Peripheral blood neutrophil granulocytes increased (fig 1),7–9 whereas peripheral blood eosinophils decreased after ACS.8 ACS has different effects on subsets of blood lymphocytes: the number of CD19 positive B cells7 and the total number of lymphocytes were depressed by ACS,8 while the number of CD3 positive cells and the CD4/CD8 ratio did not change.7 In capillary blood (finger) the total number of basophils decreased 10 minutes after smoking two cigarettes10 and the number of degranulated basophils increased.11

Increase in plasma elastase levels at 1 hour and blood neutrophil counts at 1 and 2 hours after smoking eight cigarettes in 2 hours compared with non-smoking. Reprinted from Abboud et al9 with permission.
Neutrophil kinetics in the lungs can be examined by measuring the removal of radiolabelled neutrophils during the first passage through the pulmonary circulation. MacNee et al showed increased neutrophil retention in the lungs after ACS using this method.12 This increased neutrophil retention was not due to differences in pulmonary haemodynamics,13 but may result from decreased deformability of leucocytes14 or the increased expression of the adhesion molecule l-selectin on blood neutrophils after ACS.15
Epithelial permeability as measured by 99mTc-DTPA lung clearance16 can be used to assess the disturbance of the airspace epithelial barrier. ACS increased epithelial permeability in chronic smokers after 1 hour to levels higher than in non-smokers.5 However, Gil et al17 showed no difference in epithelial permeability 15 minutes after ACS in chronic smokers. Endothelial permeability, as measured by radiolabelled urea, decreased after ACS18 but no differences could be detected when measured by PET scanning using radiolabelled transferrin.19
Oxidative stress
The acute effects of cigarette smoking on markers of oxidative stress have been analysed in exhaled air, BALF, and blood. Most studies showed an immediate increase in oxidative stress after ACS, but in several studies smoking had no effect (table S1).
Five studies have described the effects of ACS on oxidative markers in breath condensate and exhaled air. In breath condensate 8-isoprostane, a lipid peroxidation product, increased 15 minutes after ACS (fig 2)20 and hydrogen peroxide increased 30 minutes after smoke exposure.21 Exhaled nitric oxide (eNO) increased at 1 and 10 minutes22 but decreased 5 minutes after ACS in another study.23 This inconsistency probably reflects differences in eNO measurements and subject characteristics. No difference in eNO was observed at 15,23 30 and 90 minutes24 after smoking. Breath condensate levels of nitrate increased 30 minutes after ACS, but nitrite and nitrotyrosine levels did not change.24

8-isoprostane concentrations in breath condensate in healthy smokers before smoking and 15 minutes and 5 hours after smoking. Reprinted from Montuschi et al20 with permission from the American Thoracic Society.
One study5 has investigated the effects of smoking on markers of oxidative stress in BALF, showing increased superoxide release from BALF leucocytes and an increased Trolox equivalent antioxidant capacity (TEAC). This latter surprising result can be explained by the fact that the subjects studied were all chronic smokers, associated with already high BALF levels of TEAC. No difference was seen in intracellular reduced glutathione (GSH) or oxidised glutathione (GSSG) in leucocytes or in thiobarbituric acid reactive substances (TBARS) in BALF and the epithelial lining fluid (ELF).
In peripheral blood, nitrate, nitrite and cysteine levels were depressed for a short time after smoking only one cigarette.25 No difference was observed in the production of reactive oxygen intermediates from neutrophils.7 In contrast to BALF, TBARS in plasma increased26 and TEAC in plasma decreased 1 hour after smoking.5,26 Levels of F2-isoprostane, another lipid peroxidation product, did not change in plasma,27 possibly because all subjects in this study were chronic smokers and already had high F2-isoprostane levels.
Inflammatory mediators
Six studies have investigated the effects of ACS on inflammatory mediators and generally have found increased activity and recruitment of neutrophils and macrophages. In BALF, elastase activity increased6 and leukotriene B4(LTB4) release from alveolar macrophages (AMs) decreased 1 hour after smoking.28
In plasma, neutrophil elastase (NE) was increased immediately14 and 1 hour after ACS (fig 1).9 Leukotrienes B4, D4(LTD4), and E4(LTE4) increased in peripheral blood immediately and 20 minutes after ACS, and their levels were positively correlated to C3a and C5a concentrations.29 LTE4 in urine increased twofold after smoking six cigarettes.30
Acute effects of cigarette smoke in animal models
We have identified 37 studies examining the acute effects of cigarette smoke in animal models (see table S2 available online at www.thoraxjnl.com/supplemental): 31 on inflammation and six on oxidative stress.
Most studies have been performed in guinea pigs (n = 11), mice (n = 10), and rats (n = 10). Five different methods of smoke exposure were used: nose only inhalation, nose and mouth inhalation, intratracheal inhalation, inhalation by anaesthesia mask, and inhalation via a smoking chamber. The cigarette brand differed between the studies as did the amount of smoke inhaled, ranging from 3 puffs to 30 cigarettes (table S2).
Inflammatory cells
ACS predominantly increases AMs and neutrophils in animal lung tissue and BALF (table S2). In lung tissue the volume fraction of AMs in the lung parenchyma31 and the number of neutrophils in the airway wall (mucosa and outer adventitia) were increased 6 hours after ACS.31–33 The number of mast cells in the airways was also higher 6 hours after ACS.32 The opposite was true for the number of eosinophils which were decreased 6, 12, and 24 hours after smoking.32
In BALF most studies except three34–36 showed increased numbers of AMs immediately,37–39 1 hour,37,38,40 6 hours,40 8 hours,41 and 24 hours after ACS.40,42,43 The phagocytic capacity of AMs, which is important for host defence, decreased immediately after ACS38,44,45 and had returned to normal 12 hours later.38 The viability of AMs in BALF also decreased after smoking.46 The number and percentage of neutrophils in BALF were increased after 1 hour,40,47 6 hours,40,48 15 hours,49 and 24 hours.34,35,40–43,50 In contrast, four studies did not find an effect of smoke on polymorphonuclear cells (PMNs) either immediately36,37,39 or at 1 hour37,49 and 24 hours.49 This discrepancy may be explained by differences in animal species, inhalation methods, or cigarette dose. Dhami et al34 found that the number of neutrophils in mice had returned to normal after 48 hours. Both neutrophil and monocyte chemotaxis were reported to be higher 1 hour after smoke exposure than in sham exposed control animals.48
All studies but two51,52 showed increased epithelial permeability after ACS within 30 minutes32,39,53–56 and 6 hours.40 In two studies32,40 normalisation of epithelial permeability was observed after 24 hours. Two different explanations have been put forward for the enhanced permeability—damage to the epithelial cell membrane32,53,54,57 or enlargement of the spaces between the epithelial cells.54 Epithelial permeability was further increased after ibuprofen administration,53 suggesting a role for arachidonic acid metabolism.
Oxidative stress
The acute effects of smoke inhalation on markers of oxidative stress in animals have been reported in lung tissue, BALF, and blood (table S2). Most studies showed a direct increase in oxidative stress after ACS.
In lung tissue of rats the amounts of GSH decreased immediately35,58 and 1 hour after exposure to smoke.40,59 After 2–6 hours GSH levels had either returned to normal58,59 or were higher than baseline.35 GSSG levels increased at 1 hour, decreased at 6 hours, and normalised at 24 hours after ACS.40 ACS did not influence the amount of cysteine, an essential amino acid for the synthesis of GSH,59 but it increased several other markers of oxidative stress in lung tissue including 8-OhdG, 4-HNE,35,60 inducible nitric oxide synthase (iNOS) mRNA, and endothelial nitric oxide synthase (eNOS) mRNA.61
In BALF extracellular GSH was shown to be reduced immediately,59 1 hour, and 6 hours after smoke inhalation.40 After 24 hours GSH concentrations returned to baseline levels.40 ACS also depleted intracellular GSH concentrations.59 It increased GSSG36 and 8-OHdG levels60 and decreased BALF levels of TEAC.36
In blood no effect from smoke inhalation has been observed on GSH.59 However, ACS decreased the antioxidants methylumbelliferone glucuronide and ferroxidase35,62 and increased lipid peroxide and 8-epi-PGF2α, markers of lipid peroxidation in blood.36
Inflammatory mediators
The acute effects of smoke inhalation on inflammatory mediators in animals have been described in lung tissue, BALF, and blood (table S2).
In lung tissue, tumour necrosis factor α(TNF-α), macrophage inflammatory protein (MIP), and macrophage chemoattractant protein 1 (MCP-1) gene expression increased 2 hours after smoke inhalation and normalised 6 hours thereafter.42,50,63 Lung TNF-α was increased at 2, 6 and 24 hours, and e-selectin was increased at 6 and 24 hours.63
In BALF complement factor 3 increased 1 hour after ACS48 and TNF-α release from AMs was augmented after 8 hours.41 In contrast, LTB4, another important chemoattractant, decreased directly after ACS.53 Pessina et al64 showed that interleukin (IL)-6 was partially degraded after ACS.
One study showed an increase in the elastase inhibitory capacity (EIC) in BALF after ACS,49 but two other studies showed a decrease in the EIC in BALF65 and plasma.35 Furthermore, Churg et al34,42,43,50 showed a consistent increase in desmosine and hydroxyproline, both degradation products of the extracellular matrix, in BALF of smoke exposed animals after 6 and 24 hours (fig 3). The above findings suggest that acute smoke exposure can result in damaging effects on lung tissue.

Desmosine and hydroxyproline increased in bronchoalveolar lavage fluid of mice 6 and 24 hours after acute cigarette smoking. Reprinted from Dhami et al34 with permission from the American Thoracic Society.
Only two studies have been published on the effects of smoke exposure on blood inflammatory mediators, showing an increase in myeloperoxidase (MPO)66 but no changes in LTB4 levels.53
Acute effects of cigarette smoke in in vitro models
Sixty two studies examining the acute effects of cigarette smoke in in vitro models were identified (see table S3 available online at www.thoraxjnl.com/supplemental): 50 on inflammation and 12 on oxidative stress.
Many different cells and cell lines have been used in acute smoke experiments (table S3). The following cells were most frequently described: AMs (n = 12), type II alveolar epithelial cell lines (A549, n = 10) and PMNs (n = 10). The methods of cigarette smoke exposure used were different between the studies. Fifty three studies used a cigarette smoke extract (CSE) and 14 used whole cigarette smoke (CS). The concentration of CSE and the time of exposure differed considerably between the studies with concentrations varying from 8 × 10−5 cigarette/ml to 4 cigarette/ml and exposure times varying between 1 second and 24 hours, respectively.
Inflammatory cells
In vitro studies have shown various effects of CS and CSE on different cell characteristics which may provide useful information to enable a better understanding of the effects of smoking in vivo. Neutrophil and monocyte chemotactic activity of the supernatant of epithelial cells and fibroblasts incubated in CSE for 3–24 hours increased.67–69 This increase diminished after lipoxygenase inhibitors and arachidonic acid metabolite inhibitors had been added.67–69 In contrast, the chemotactic response of blood PMNs exposed directly to CS or CSE appeared to be decreased70 or unchanged.71 This suggests that CSE has an indirect effect on PMN chemotaxis.
Adhesion of human PMNs to a type II alveolar epithelial cell line decreased directly after exposure to CS,71 but adhesion of human PMNs to a primary bovine bronchial epithelial cell line (BBEC) increased after incubation in CSE for 24 hours.72 The adhesion of human monocytes to human umbilical vein endothelial cells (HUVEC) and human bronchial epithelial cells (HBEC) was also increased when incubated in CSE.73,74 This might result from an increased expression of adhesion molecules CD11b, intercellular adhesion molecule 1 (ICAM-1), endothelial leucocyte adhesion molecule 1 (ELAM-1), and vascular cell adhesion molecule 1 (VCAM-1).73,75 The expression of CD18 on human PMNs was increased in one study76 but remained unchanged in another.71 Surprisingly, ACS decreased the expression of l-selectin on PMNs.76
The phagocytic capacity of AMs, peritoneal macrophages (PMs), and PMNs was shown to decrease during CS exposure and 30 minutes, 2 and 24 hours after exposure to CS.77–80 Increased phagocytic capacity of mice AMs was seen after exposure to only a low dose of CS.77 The protein synthesis of rabbit AMs was depressed directly after CSE exposure and was restored after 24 hours.81,82
ACS can affect the function of fibroblasts in vitro. CSE inhibited the proliferation of human fetal lung fibroblasts (HFL1),83 decreased fibronectin release,84,85 viability and protein synthesis of fibroblasts,81,86 and depressed fibroblast collagen mediated gel contraction, a model for wound repair.84,85
The viability of alveolar epithelial cells and AMs and PMs decreased after ACS in a concentration and time dependent manner.46,77,79,87 Primary murine fibroblasts were less susceptible to cell death induced by CSE than murine AMs.46 Six studies have shown that CSE resulted in apoptosis within 3–24 hours in different cell types.86,88–92 However, Wickenden et al93 showed that CSE exposure only induced necrosis. This might partly be explained by the fact that different cell types and CSE concentrations were used. Interestingly, two studies showed that exposing cells to low concentrations of CSE induced apoptosis while high concentrations resulted in necrosis.91,92
Two studies94,95 on epithelial permeability in vitro showed an increase at 20 minutes and 1 hour after exposure to CS and CSE. Glutathione reduced this effect,95 suggesting that oxidants contribute to the increase in epithelial permeability. Other interesting acute effects of CSE have been found. Firstly, CSE inhibited surfactant secretion of alveolar type II cells after 20 minutes of exposure.96 Pinot et al97 showed that surfactant can prevent oxidative stress induced by CSE in vitro. These results have clinical relevance since surfactant is important in maintaining alveolar stability and plays a role in alveolar and also (though less prominently) in bronchial clearance. Secondly, Takeyama et al98 showed that CSE increased mucin synthesis by a pulmonary mucoepidermoid cell line already within 24 hours. This suggests the possibility of a rapid upregulatory mechanism of mucus production in vivo in chronic smokers. A decrease in mucus flow on ciliated epithelium was seen within minutes of exposure to CS.99
Oxidative stress
Twelve studies have investigated the effect of ACS on oxidative stress, all showing an increase in oxidative stress after exposure to CS. GSSG was released after 30 minutes100 and intracellular GSH was decreased within 3 hours of ACS exposure.86,95,101 When measurements were performed 24 hours after exposure, GSH and γ-GCS were in fact increased, suggesting a protective mechanism of cells against oxidative stress from smoke.102 Immediately after six puffs of smoke, hydrogen peroxide and superoxide molecules from CS were detectable along the membranes of epithelial cells,103 which were prevented by antioxidants. After 24 hours of incubation with CSE, nitric oxide (NO) was released from endothelial cells.88 In contrast, iNOS expression and nitrate release from stimulated epithelial cells were decreased after CSE exposure.104 The pentose phosphate pathway, the source of NADPH for the enzyme gluthatione reductase, was activated after incubation of endothelial cells with CSE.100
Inflammatory mediators
All studies but one105 showed an increased release of IL-8 in various cell types after different exposure times to CSE (20 minutes in HBEC,105 4 and 8 hours in human endothelial cells,106 6 hours mRNA IL-8 in NCI-H292,107 12 hours in HBEC,108 and 24 hours in HBEC and A549 cell line108,109). The results of the two negative studies might be explained by the low concentrations of CSE, the use of CS instead of CSE, or by the different cell types used.
Inconsistent results were also found for IL-1β, TNF-α, and soluble ICAM (sICAM): IL-1β and sICAM were increased in HBEC 20 minutes, 1 hour and 24 hours after exposure to CS94,105 but were decreased when HBEC were exposed for 3 and 6 hours.105 IL-1β and TNF-α release was increased when peripheral blood mononuclear cells (PBMCs) were exposed for 5 minutes110 but decreased after 3 hours exposure.111 TNF-α release from AMs was decreased when exposed for 1 hour at low concentrations112 but increased when exposed for 18 hours with higher concentrations of CSE.63 CSE had no effect on sICAM release from HUVEC at 24 hours.113 mRNA expression of IL-8, IL-1β, and sICAM was increased after 30 minutes of incubation of HBEC in CSE.114
Cigarette smoke has been shown to have a depressive effect on some other inflammatory mediators in vitro. The release of LTB4 from AMs28 and interferon-γ(IFN-γ) and IL-2111 from human PBMCs was less after incubation in CSE. The activity of both IL-6 and TNF-α secreted by AMs was diminished after exposure to CS.115 CSE had no direct effect on the release of NE from human blood PMNs in vitro.116
DISCUSSION
Smoking is the main risk factor for the accelerated decline in lung function and development of COPD. Much is known of the effects of chronic smoke exposure on lung function and airway inflammation, but there is a paucity of data on the acute effects of smoking in this respect. It seems important to know these effects since repetitive acute smoke effects may constitute the underlying causal chain leading to the ultimate chronic effects.
We have identified 123 studies investigating the acute effects of CS on inflammatory cells, oxidative stress, and inflammatory mediators in humans, animals and in vitro models. Various cigarette brands with and without a filter and different doses have been studied, ranging from 1 puff to 30 cigarettes. Different time points and several body compartments in humans and animals have been investigated. An extensive collection of information has therefore been acquired, yet of various natures.
One of the problems in the comparison of the various studies is the difference in the way human, animal, and in vitro models have been exposed to smoke. Firstly, even though animals have a much smaller lung surface than humans, this review shows that animals are exposed to a higher number of cigarettes than humans (median 5 cigarettes (range 0.9–34)v median 2 cigarettes (range 1–24)). Secondly, in vitro studies mainly used CSE whereas all humans and almost all animals were exposed to CS. The composition of CSE and CS has important differences, especially regarding the water insoluble substances and free radicals.117–119 Thus, the results of different models cannot therefore simply be compared.
In this review we have provided data that are of interest and importance to the damaging effects of smoke in diseases in general. We have shown that ACS is chemotactic to neutrophils and macrophages and activates these cells. Furthermore, acute smoke exposure results in tissue damage, as suggested by increased products of lipid peroxidation and matrix degradation products. A very intriguing finding was the suppressive effect of ACS on the number of eosinophils and several inflammatory cytokines. It may well be that this suppressive effect results from the anti-inflammatory carbon monoxide (CO) present in cigarette smoke or produced by inflammatory cells in the lung.120
Inflammatory cells
This review shows that neutrophils are already attracted and activated after the first puffs of CS in both human and animal studies. In line with this, increased neutrophil chemotactic activity of supernatant of epithelial cells exposed to CS was observed in vitro.
ACS induces increased numbers of AMs in animal lung tissue and BALF, but not in human BALF. This may be due to the short time interval or the low dose of smoke used. Furthermore, increased monocyte chemotactic activity of BALF and supernatant of epithelial cells exposed to CS was observed. Eosinophils seem to play a role in a subgroup of patients with stable COPD121 and in those with COPD exacerbations.122 ACS directly increased eosinophil numbers in animal BALF.37 Intriguingly, two other studies8,32 have shown a suppressive effect of smoke on the number of eosinophils in human blood and in animal tissue. This may be a reflection of local shifts in the Th1–Th2 type cytokine balance or an anti-inflammatory effect of substances in smoke such as CO.123,124
The effect of ACS on apoptosis and necrosis has mainly been investigated in in vitro studies. Interestingly, two studies showed that exposure of cells to low concentrations of CSE induced apoptosis but high concentrations of CSE resulted in necrosis.91,92 Because apoptosis of (inflammatory) cells is associated with less damage of the extracellular matrix, one might even hypothesise that smokers who smoke intermittently or only a few cigarettes per day are less likely to develop lung damage than those who smoke many cigarettes in a chain.
ACS increased the air space epithelial permeability in human, animal, and in vitro studies. This increase was shown to occur within an hour after exposure to CS and returned to normal within 24 hours. Theoretically, impairment of the epithelial barrier may potentiate the damaging effects of noxious agents in the lung.
ACS also inhibits the function of fibroblasts which are important in repair processes in the lung. Injury and repair processes of the airway epithelium have been studied extensively in chronic airway disease. It is assumed that these repeated injury and repair processes may contribute to the development of airway pathology in chronic inflammatory airway diseases.125 Repetition of acute smoke exposure may lead in this way to irreversible damage, especially if fibroblasts are not functioning normally. More studies on this subject should be performed to strengthen this hypothesis.
Summarising, ACS increases local inflammation as reflected by an increase in the number of neutrophils and macrophages in the lung. It reduces important qualitative cell characteristics, repair mechanisms, and the protection of the epithelial barrier. Furthermore, ACS results in a decrease in the number of eosinophils, indicating a possible local shift in the Th1–Th2 type cytokine balance or an anti-inflammatory effect of CO.
Oxidative stress
ACS increases markers of oxidative stress in all three models (human, animal, and in vitro). NO and GSH are the only two parameters that have been investigated in all models. NO and its related substances increase within 24 hours after smoke exposure. The GSH/GSSG ratio, reflecting the vital balance between oxidants and protecting antioxidants, decreased following acute smoke exposure in both animal and in vitro studies but not in the single study published in humans. This discrepancy can be explained by differences in species, smoke dose, or compartment (human BALF versus animal lung homogenate).
Interestingly, ACS even results in damage of fatty acids in cell membranes, as measured by an increase in degradation products of lipid peroxidation in humans (exhaled air and plasma)20,26 and animals (BALF and lung tissue).35,60 No in vitro studies investigating the acute smoke effects on lipid peroxidation products have been found.
Because different time points within 24 hours have been studied, it allowed us to observe a time response of oxidative stress. In humans all oxidative markers increase within the first hour after ACS and most markers returned to normal within 90 minutes. Exhaled air is the first compartment in which an increase in oxidative stress markers can be observed, followed by BALF and blood. In animals most markers of oxidative stress change in the first 6 hours after ACS and return to normal within 24 hours. In all compartments (lung tissue, BALF, and blood) GSH or its derivatives are depressed in the same time period, suggesting a generalised response to ACS. As in humans, only a few time points have been studied in in vitro models. The initial depletion of GSH after ACS appeared to be followed by an increase in GSH 24 hours later, suggesting a protective mechanism of cells against oxidative stress from smoke.102 The importance of the GSH/GSSG balance was shown in several studies. When GSH was added to the experiment the oxidative stress and inflammatory response induced by cigarette smoke could be prevented.
In summary, ACS immediately increases markers of oxidative stress in all models and even results in damage to the cell membrane. The GSH/GSSG balance plays an important role in the acute protection of the lung against oxidants in CS.
Inflammatory mediators
ACS induces a wide range of (pro)inflammatory responses. All three models (human, animal, and in vitro) studied the effect of ACS on NE, leukotrienes, and IL-6. Interestingly, NE was released only a few hours after a low dose of CS, both in animals and in humans. In contrast, direct exposure of human PMNs in vitro for 4 minutes did not affect the release of NE. This suggests that CS does not affect NE release by neutrophils directly, indicating that the local microenvironment may have a role in mounting this response. Another explanation might be that the in vitro exposure time was too short to activate these cells.
Inconsistent results have been shown for the effects of ACS on leukotrienes, with increased (human, in vitro), decreased (animal, in vitro), or no effects (animal). This could be due to differences in cigarette dose, cell type, or species under study.53
IL-6, which plays a role in innate and adaptive immunity, was also studied in all models. Alveolar macrophage IL-6 activity was decreased after in vitro smoke exposure and IL-6 degradation was increased in BALF of rats.64,115 No effect of ACS was found on human blood levels of IL-6,7 suggesting that ACS may have a depressive effect only locally in the bronchial tree or that is compensated for by IL-6 production by other cells.
In vitro, ACS increased the release of IL-8 from epithelial and endothelial cells and cell lines. This is in line with the observed increase in neutrophils after ACS in humans and animals, which suggests that IL-8 is a chemoattractant for neutrophils after exposure to ACS.
A suppressive effect of ACS was seen in some inflammatory mediators (TNF-α, IFN-γ, LTB4, and IL-2) in vitro.28,111,112,115 This suppressive effect may result from CO from CS or is produced by heme oxygenase-1 (HO-1) in inflammatory cells in the lung.120
In summary, ACS can disturb the balance between proteases such as NE and their inhibitors, possibly resulting in early tissue damage. In addition, it increases IL-8 which may contribute to chemotaxis of neutrophils as found after ACS. Interestingly, ACS has a suppressive effect on some inflammatory mediators, possibly due to the anti-inflammatory effect of CO.
Susceptible smoker
A vital question when investigating the development of COPD is how to pinpoint the susceptible smoker. Differences in smoke exposure and genetic factors do not give the complete answer. In this review we describe an acute decrease in the GSH/GSSG ratio after smoke exposure. This decrease puts the smoker at risk to oxidants of CS soon after the first exposure. The extent and velocity to which the GSH/GSSG balance is restored probably determines to some extent the degree of susceptibility. The balance between proteases and antiproteases may also have a role, but studies performed to date have shown contradictory results. One study showed that NE and EIC in animal BALF increase simultaneously after smoke exposure, suggesting a protective mechanism. Yet, acute smoke exposure in three other studies showed an increase in the matrix degradation products desmosine and hydroxyproline in animal BALF. This supports the hypothesis that the ability to maintain the balance between proteases and antiproteases is of vital importance for protecting the lung against proteolysis. Finally, a polymorphism in the HO-1 promoter region has been described in patients with COPD, resulting in a lower production of HO-1.126 This review shows that ACS decreases the number of eosinophils and some inflammatory mediators which might be caused by the anti-inflammatory CO produced locally by HO-1 in the lung. One might hypothesise that, in smokers, HO-1 expression is important for the susceptibility to develop COPD. More studies on acute smoking with larger groups should be performed to further unravel this complicated but very important issue.
CONCLUSIONS
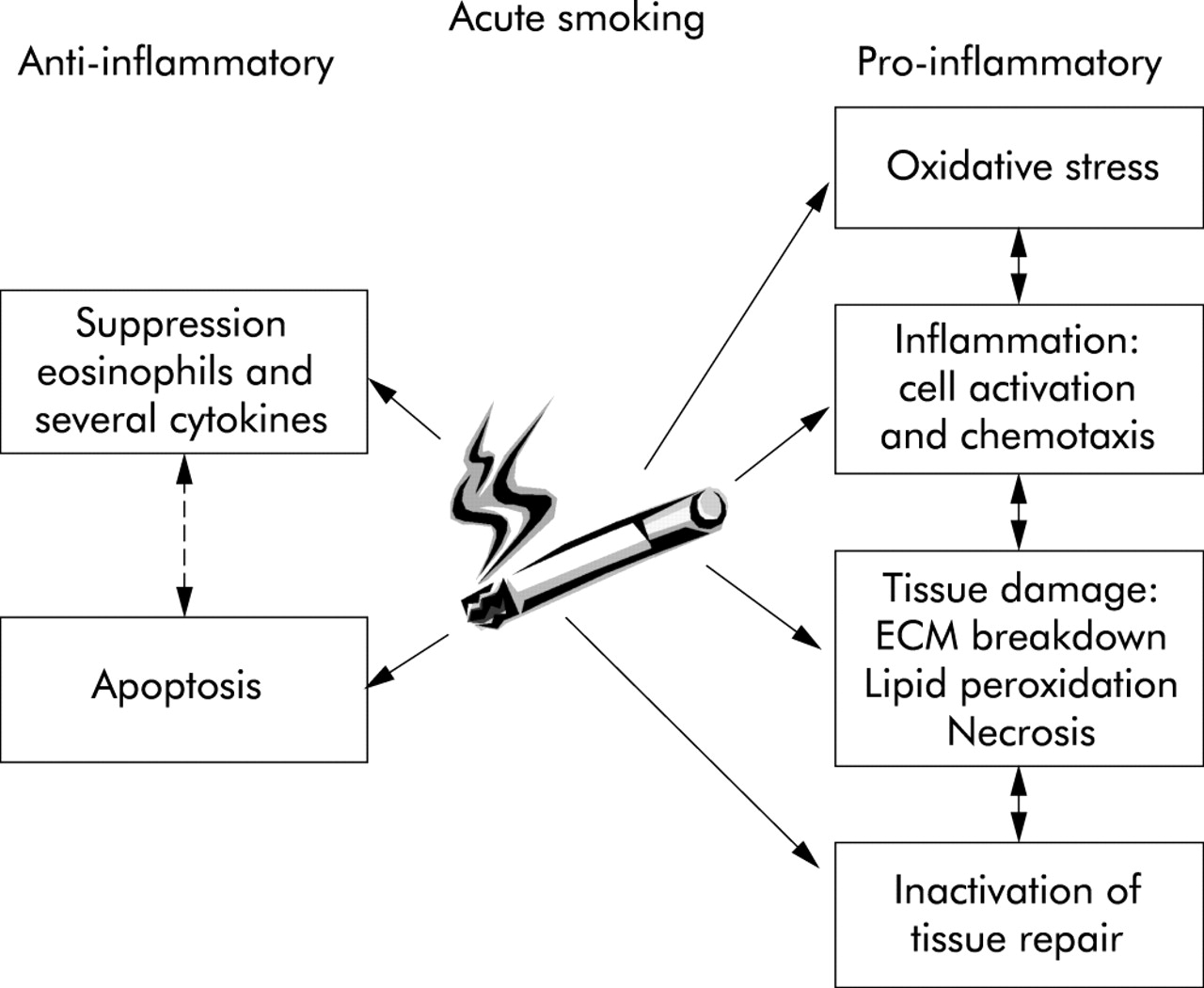
This review shows that an acute smoking model is a relatively easy and sensitive method for investigating the specific effects of cigarette smoke on oxidative stress and inflammation. We have shown that ACS is chemotactic to neutrophils and macrophages and activates these cells. An intriguing finding was the suppressive effect of ACS on the number of eosinophils and several inflammatory cytokines, possibly explained by a local shift in the Th1–Th2 type cytokine balance or by the anti-inflammatory effect of CO. Importantly, even acute smoke exposure might result in tissue damage, as suggested by increased products of lipid peroxidation and degradation products of extracellular matrix proteins. This review supports the view that an imbalance between oxidants and antioxidants and between proteases and antiproteases may play an important role in the susceptible smoker, and it has become clear that disturbances in effective tissue repair also deserve attention (fig 4). It is, however, difficult to draw firm conclusions because of the small sample sizes studied, essential differences between human, animal and in vitro models, and other methodological divergences. An acute smoking model is a useful supplement to other methods of studying the effects of smoking, and is an as yet underinvestigated method for intervention studies in smoking related diseases such as COPD.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of the acute effects of cigarette smoking. Data extracted from human, animal, and in vitro studies. ECM, extracellular matrix.
REFERENCES
Supplementary materials
Web-only Tables
These tables are available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
- [View PDF] -
Table S1
Twenty five studies examining the acute effects of cigarette smoking (ACS) in humansTable S2
37 studies examining the acute effects of cigarette smoke in animal modelsTable S3
Sixty two studies examining the acute effects of cigarette smoke in in vitro models
- [View PDF] -
Footnotes
-
This review was supported by a grant from Astra Zeneca.